Wang-yang Wu, Zong-xiang Zheng, Hao-bo Wan, Jie Mao, Fang-li Wang, Lei Yang, Mohamad Akbar Ali, Ling-hai Xie
{"title":"Optoelectronic behavior of spirocyclopentadithiophene in lattice aromatics","authors":"Wang-yang Wu, Zong-xiang Zheng, Hao-bo Wan, Jie Mao, Fang-li Wang, Lei Yang, Mohamad Akbar Ali, Ling-hai Xie","doi":"10.1007/s00894-025-06411-x","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>This study investigates the influence of intramolecular π-π stacking interactions on the optoelectronic properties of spirocyclopentadithiophene (spiro-CPDT)-based latticed molecules (GS-CPDT, HGS-CPDT1, and HGS-CPDT2) to optimize their charge transport characteristics. Density functional theory was employed to analyze molecular geometries, frontier molecular orbitals, adiabatic ionization potentials (<i>IP</i><sub><i>a</i></sub>), electron affinities (<i>EA</i><sub><i>a</i></sub>), and reorganization energies (<i>λ</i>). Crystal structure modeling using the Dreiding force field and computational evaluation of electronic coupling parameters (<i>V</i><sub><i>e</i></sub>, <i>V</i><sub><i>h</i></sub>) and charge-transfer rate constants (<i>k</i><sub><i>e</i></sub>, <i>k</i><sub><i>h</i></sub>) were performed to assess intramolecular π-π stacking effects. Results reveal that lattice-induced π-stacking configurations significantly reduce reorganization energies (<i>λ</i><sub><i>e</i></sub> = 0.223 eV, <i>λ</i><sub><i>h</i></sub> = 0.343 eV) while enhancing charge-transfer rate constants (~ 10<sup>11</sup> s⁻<sup>1</sup>), demonstrating improved charge transport efficiency compared to conventional spiro-CPDT systems. These findings establish fundamental structure–property relationships for spiro-aromatic hydrocarbons, offering critical theoretical guidance for designing organic electronic materials with tailored charge transport capabilities.</p><h3>Methods</h3><p>The molecular energy, molecular structure, molecular orbitals, and other properties of all molecules designed in this paper were calculated using functional B3LYP and basis set 6-31G(d). Based on these calculations, tasks such as optimizing the ground state geometry of the molecules, calculating electrostatic potential, and optoelectronic properties were carried out. The weak interactions of molecules were investigated using Multiwfn 3.8 and VMD. Finally, the molecular crystal structure was predicted using the Metamorph module in Materials Studio 2020, and the dimer of the studied molecule was obtained.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 7","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-06-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06411-x","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
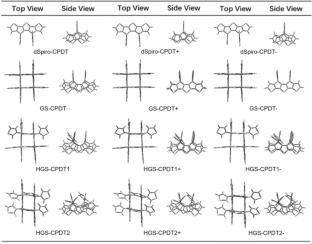
This study investigates the influence of intramolecular π-π stacking interactions on the optoelectronic properties of spirocyclopentadithiophene (spiro-CPDT)-based latticed molecules (GS-CPDT, HGS-CPDT1, and HGS-CPDT2) to optimize their charge transport characteristics. Density functional theory was employed to analyze molecular geometries, frontier molecular orbitals, adiabatic ionization potentials (IPa), electron affinities (EAa), and reorganization energies (λ). Crystal structure modeling using the Dreiding force field and computational evaluation of electronic coupling parameters (Ve, Vh) and charge-transfer rate constants (ke, kh) were performed to assess intramolecular π-π stacking effects. Results reveal that lattice-induced π-stacking configurations significantly reduce reorganization energies (λe = 0.223 eV, λh = 0.343 eV) while enhancing charge-transfer rate constants (~ 1011 s⁻1), demonstrating improved charge transport efficiency compared to conventional spiro-CPDT systems. These findings establish fundamental structure–property relationships for spiro-aromatic hydrocarbons, offering critical theoretical guidance for designing organic electronic materials with tailored charge transport capabilities.
Methods
The molecular energy, molecular structure, molecular orbitals, and other properties of all molecules designed in this paper were calculated using functional B3LYP and basis set 6-31G(d). Based on these calculations, tasks such as optimizing the ground state geometry of the molecules, calculating electrostatic potential, and optoelectronic properties were carried out. The weak interactions of molecules were investigated using Multiwfn 3.8 and VMD. Finally, the molecular crystal structure was predicted using the Metamorph module in Materials Studio 2020, and the dimer of the studied molecule was obtained.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


