{"title":"Evaluation of thermochemical characteristics of salts with pentazenium cation","authors":"D. V. Khakimov, A. A. Voronin","doi":"10.1007/s00894-025-06420-w","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>Using quantum-chemical and crystal modeling, the solid-state enthalpies of three hypothetical high-energy salts with the pentazenium cation N<sub>5</sub><sup>+</sup> were estimated: nitrate NO<sub>3</sub><sup>−</sup>, dinitramide N(NO<sub>2</sub>)<sub>2</sub><sup>−</sup>, and azide N<sub>3</sub><sup>−</sup>, yielding the cationic contribution of pentazenium. Supplementing the lattice energy mixing method with an additive approach to determining the enthalpies of salts, values are given for salts with the perchlorate anion N<sub>5</sub><sup>+</sup>ClO<sub>4</sub><sup>−</sup>; four salts with the halogen anions N<sub>5</sub><sup>+</sup>I<sup>−</sup>, N<sub>5</sub><sup>+</sup>Br<sup>−</sup>, N<sub>5</sub><sup>+</sup>Cl<sup>−</sup>, and N<sub>5</sub><sup>+</sup>F<sup>−</sup>; and two experimentally existing structures: pentazenium tetrafluoroborate N<sub>5</sub><sup>+</sup>BF<sub>4</sub><sup>−</sup> and pentazenium hexafluorophosphate N<sub>5</sub><sup>+</sup>PF<sub>6</sub><sup>−</sup>. Calculations of salt structures were performed using a wide range of density functional theory (DFT) methods with varying functionals and bases, as well as composite methods for calculating the enthalpies of gas phase formation, to show the low variability of the final result and the universality of the approach. The calculations of the explosive characteristics of salts with oxygen–nitrogen anions showed that their detonation velocity is in the range of 7.1–7.6 km s<sup>−1</sup>. Of the three salts considered, the azide salt has the lowest density equal to 1.1 g cm<sup>−3</sup>, while the nitrate and dinitramide are at the level of 1.6 g cm<sup>−3</sup> and 1.7 g cm<sup>−3</sup>, respectively. The heat of detonation of pentazenium salts is about 800–850 cal g<sup>−1</sup>.</p><h3>Methods</h3><p>In this work, broad set of DFT calculations were conducted through the software Gaussian 09: B3LYP/6-31G(d,p), B3LYP/aug-cc-PVDZ + GD2, M052X/aug-cc-pVTZ, and M062X/6–311 + + G(d,p). For crystal structure optimization, the atom–atom potential methods with PMC program (Packing of Molecules in Crystal) were used. Charges for molecular electrostatic potential were fitted by FitMEP, and enthalpies of formation in gas phase were assessed by CBS-4 M, G3B3, G4, and W1BD.\n</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"31 7","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2025-06-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-025-06420-w","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
Using quantum-chemical and crystal modeling, the solid-state enthalpies of three hypothetical high-energy salts with the pentazenium cation N5+ were estimated: nitrate NO3−, dinitramide N(NO2)2−, and azide N3−, yielding the cationic contribution of pentazenium. Supplementing the lattice energy mixing method with an additive approach to determining the enthalpies of salts, values are given for salts with the perchlorate anion N5+ClO4−; four salts with the halogen anions N5+I−, N5+Br−, N5+Cl−, and N5+F−; and two experimentally existing structures: pentazenium tetrafluoroborate N5+BF4− and pentazenium hexafluorophosphate N5+PF6−. Calculations of salt structures were performed using a wide range of density functional theory (DFT) methods with varying functionals and bases, as well as composite methods for calculating the enthalpies of gas phase formation, to show the low variability of the final result and the universality of the approach. The calculations of the explosive characteristics of salts with oxygen–nitrogen anions showed that their detonation velocity is in the range of 7.1–7.6 km s−1. Of the three salts considered, the azide salt has the lowest density equal to 1.1 g cm−3, while the nitrate and dinitramide are at the level of 1.6 g cm−3 and 1.7 g cm−3, respectively. The heat of detonation of pentazenium salts is about 800–850 cal g−1.
Methods
In this work, broad set of DFT calculations were conducted through the software Gaussian 09: B3LYP/6-31G(d,p), B3LYP/aug-cc-PVDZ + GD2, M052X/aug-cc-pVTZ, and M062X/6–311 + + G(d,p). For crystal structure optimization, the atom–atom potential methods with PMC program (Packing of Molecules in Crystal) were used. Charges for molecular electrostatic potential were fitted by FitMEP, and enthalpies of formation in gas phase were assessed by CBS-4 M, G3B3, G4, and W1BD.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.
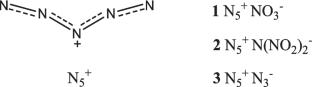

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


