{"title":"A compact encoding of the genome suitable for machine learning prediction of traits and genetic risk scores.","authors":"Yasaman Fatapour, James P Brody","doi":"10.1186/s13040-025-00459-4","DOIUrl":null,"url":null,"abstract":"<p><p>Genotype to phenotype prediction is a central problem in biology and medicine. Machine learning is a natural tool to address this problem. However, a person's genotype is usually represented by a few million single-nucleotide polymorphisms and most datasets only have a few thousand patients. Thus, this problem typically has many more predictors than the number of samples (patients), making it unsuitable for machine learning. The objective of this paper is to examine the efficacy of a compact genotype representation, which employs a limited number of predictors, in predicting a person's phenotype through the application of machine learning. We characterized a person's genotype using chromosome-scale length variation, a measure that is computed as the average value of reported log R ratios across a portion of a chromosome. We computed these numbers from data collected by the NIH All of Us program. We used the AutoML function (h2o.ai) in binary classification mode to identify the best models to differentiate between male/female, Black/white, white/Asian, and Black/Asian. We also used the AutoML function in regression mode to predict the height of people based on their age and genotype. Our results showed that we could effectively classify a person, using only information from chromosomes 1-22, as Male/Female (AUC = 0.9988 ± 0.0001), White/Black (AUC = 0.970 ± 0.002), Asian/White (AUC = 0.877 ± 0.002), and Black/Asian (AUC = 0.966 ± 0.002). This approach also effectively predicted height. In conclusion, we have shown that this compact representation of a person's genotype, along with machine learning, can effectively predict a person's phenotype.</p>","PeriodicalId":48947,"journal":{"name":"Biodata Mining","volume":"18 1","pages":"44"},"PeriodicalIF":6.1000,"publicationDate":"2025-06-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12180147/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Biodata Mining","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13040-025-00459-4","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
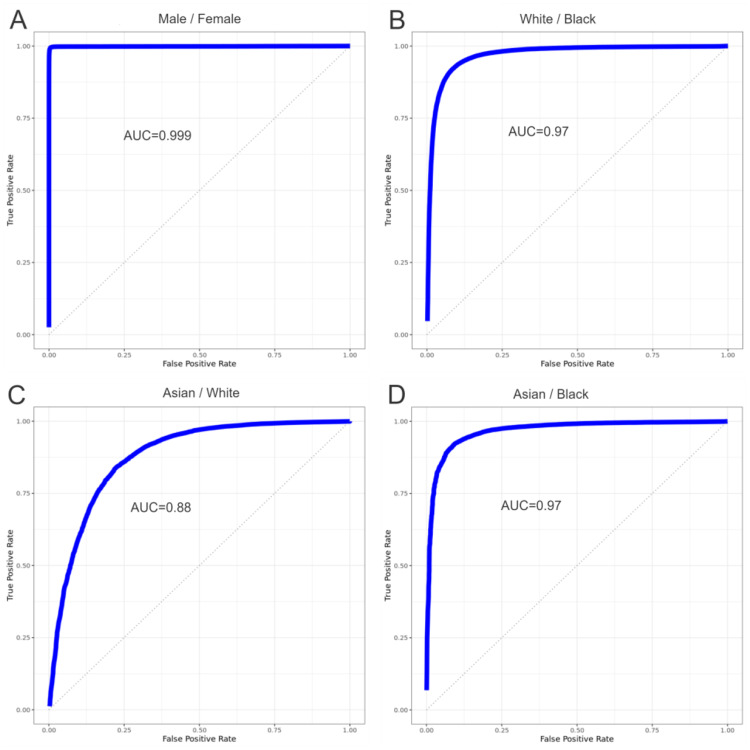
Genotype to phenotype prediction is a central problem in biology and medicine. Machine learning is a natural tool to address this problem. However, a person's genotype is usually represented by a few million single-nucleotide polymorphisms and most datasets only have a few thousand patients. Thus, this problem typically has many more predictors than the number of samples (patients), making it unsuitable for machine learning. The objective of this paper is to examine the efficacy of a compact genotype representation, which employs a limited number of predictors, in predicting a person's phenotype through the application of machine learning. We characterized a person's genotype using chromosome-scale length variation, a measure that is computed as the average value of reported log R ratios across a portion of a chromosome. We computed these numbers from data collected by the NIH All of Us program. We used the AutoML function (h2o.ai) in binary classification mode to identify the best models to differentiate between male/female, Black/white, white/Asian, and Black/Asian. We also used the AutoML function in regression mode to predict the height of people based on their age and genotype. Our results showed that we could effectively classify a person, using only information from chromosomes 1-22, as Male/Female (AUC = 0.9988 ± 0.0001), White/Black (AUC = 0.970 ± 0.002), Asian/White (AUC = 0.877 ± 0.002), and Black/Asian (AUC = 0.966 ± 0.002). This approach also effectively predicted height. In conclusion, we have shown that this compact representation of a person's genotype, along with machine learning, can effectively predict a person's phenotype.
期刊介绍:
BioData Mining is an open access, open peer-reviewed journal encompassing research on all aspects of data mining applied to high-dimensional biological and biomedical data, focusing on computational aspects of knowledge discovery from large-scale genetic, transcriptomic, genomic, proteomic, and metabolomic data.
Topical areas include, but are not limited to:
-Development, evaluation, and application of novel data mining and machine learning algorithms.
-Adaptation, evaluation, and application of traditional data mining and machine learning algorithms.
-Open-source software for the application of data mining and machine learning algorithms.
-Design, development and integration of databases, software and web services for the storage, management, retrieval, and analysis of data from large scale studies.
-Pre-processing, post-processing, modeling, and interpretation of data mining and machine learning results for biological interpretation and knowledge discovery.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


