Clinicopathological and Immunogenetic Characterization in 8 Patients with Familial Hemophagocytic Lymphohistiocytosis Type 2: A Study from North India with Literature Review.
{"title":"Clinicopathological and Immunogenetic Characterization in 8 Patients with Familial Hemophagocytic Lymphohistiocytosis Type 2: A Study from North India with Literature Review.","authors":"Saniya Sharma, Suprit Basu, Taru Goyal, Madhubala Sharma, Prabal Barman, Gurjit Kaur, Jitendra K Shandilya, Pandiarajan Vignesh, Rakesh Kumar Pilania, Ankur Kumar Jindal, Manpreet Dhaliwal, Prateek Bhatia, Sreejesh Sreedharanunni, Pulkit Rastogi, Nabhajit Mallik, Prashant Sharma, Anupriya Kaur, Deepti Suri, Amit Rawat, Surjit Singh","doi":"10.1007/s10875-025-01895-x","DOIUrl":null,"url":null,"abstract":"<p><p>Familial hemophagocytic lymphohistiocytosis type 2 (FHL2) is the commonest cause of familial hemophagocytic lymphohistiocytosis (FHLH). In this retrospective study, we analyzed 8 patients with a genetic diagnosis of FHL2 and then examined their clinicopathological and perforin flow cytometry results (< 10% expression). The atypical clinical features in our cohort included tuberculosis, lymphoreticular malignancy, and necrotizing enterocolitis in 3 patients. A disease-causing variant was identified in the PRF1 gene in all eight patients, comprising missense (n = 6), null (n = 1), and in-frame deletion (n = 1). Five patients had homozygous exon 3 disease-causing variants, two had homozygous exon 2 disease-causing variants, and one had compound heterozygous disease-causing variants in exon 2 and exon 3. After an extensive literature search, the mutations present in our North Indian cohort, including c.1284G > A, c.895C > T, c.853_855del, c.203G > A, and c.757G > A, are reported for the first time from India. Clinical and immunological phenotypes of c.1284G > A and c.203G > A variants have not been published in the literature. Hemophagocytosis was evident in bone marrow in 6 cases. Hyperferritinemia was absent in 3 cases, including c.148G > A, c. 895C > T, and c.1349C > T homozygous variants. Neurological involvement, lymphoreticular malignancy, and necrotizing enterocolitis were seen in 2, 1, and 1 cases, respectively. Infections were present in 4 cases. Five children succumbed to HLH, and three are alive and planned for a hematopoietic stem cell transplant. FHL2 should be suspected in children with HLH irrespective of the age of onset, atypical clinical phenotype, family history, ferritin and fibrinogen levels, and infections. Flow cytometry-based perforin assay helps in rapid diagnosis of FHL2.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"45 1","pages":"108"},"PeriodicalIF":5.7000,"publicationDate":"2025-06-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12178966/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-025-01895-x","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
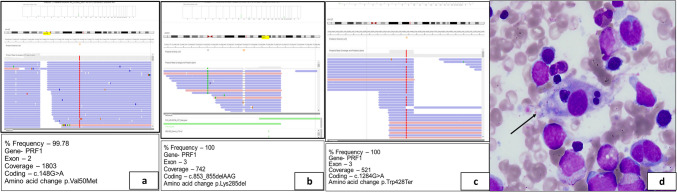
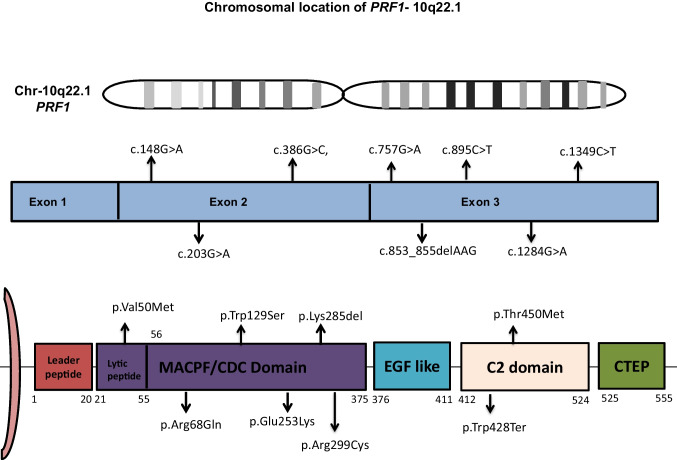
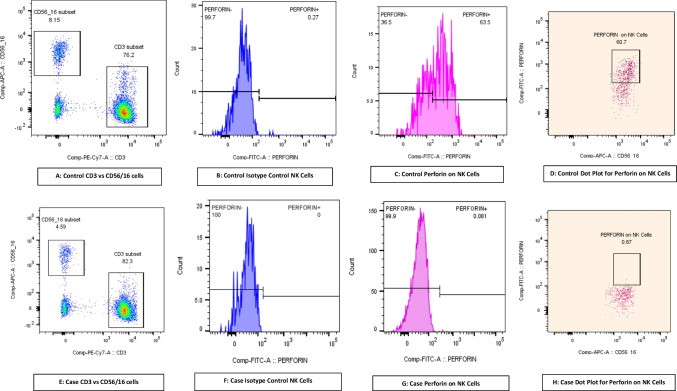
Familial hemophagocytic lymphohistiocytosis type 2 (FHL2) is the commonest cause of familial hemophagocytic lymphohistiocytosis (FHLH). In this retrospective study, we analyzed 8 patients with a genetic diagnosis of FHL2 and then examined their clinicopathological and perforin flow cytometry results (< 10% expression). The atypical clinical features in our cohort included tuberculosis, lymphoreticular malignancy, and necrotizing enterocolitis in 3 patients. A disease-causing variant was identified in the PRF1 gene in all eight patients, comprising missense (n = 6), null (n = 1), and in-frame deletion (n = 1). Five patients had homozygous exon 3 disease-causing variants, two had homozygous exon 2 disease-causing variants, and one had compound heterozygous disease-causing variants in exon 2 and exon 3. After an extensive literature search, the mutations present in our North Indian cohort, including c.1284G > A, c.895C > T, c.853_855del, c.203G > A, and c.757G > A, are reported for the first time from India. Clinical and immunological phenotypes of c.1284G > A and c.203G > A variants have not been published in the literature. Hemophagocytosis was evident in bone marrow in 6 cases. Hyperferritinemia was absent in 3 cases, including c.148G > A, c. 895C > T, and c.1349C > T homozygous variants. Neurological involvement, lymphoreticular malignancy, and necrotizing enterocolitis were seen in 2, 1, and 1 cases, respectively. Infections were present in 4 cases. Five children succumbed to HLH, and three are alive and planned for a hematopoietic stem cell transplant. FHL2 should be suspected in children with HLH irrespective of the age of onset, atypical clinical phenotype, family history, ferritin and fibrinogen levels, and infections. Flow cytometry-based perforin assay helps in rapid diagnosis of FHL2.
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


