Srilatha Arra, Isabella Daidone, Massimiliano Aschi
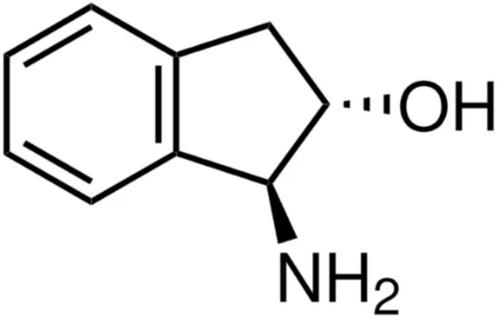
{"title":"Revisiting the “Cluster-In-Solvent” Approach for Computational Spectroscopy: The Vibrational Circular Dichroism as a Test Case","authors":"Srilatha Arra, Isabella Daidone, Massimiliano Aschi","doi":"10.1002/jcc.70144","DOIUrl":null,"url":null,"abstract":"<p>The cluster-in-solvent approach, that is, the use of the quantum-mechanical calculation of a spectral observable on a significant number of solute–solvent clusters extracted from semi-classical simulations, is widely used in computational spectroscopy. However, identifying relevant coordinates for cluster selection remains a challenge. We previously developed the Ellipsoid Method for Cluster-in-Solvent (EMCS), an automated strategy for unbiased identification and statistical weighting of clusters. Yet, for larger solutes, EMCS can yield overly large solvent clusters that hinder conformational analysis. Here, we introduce a simple extension of EMCS that reduces cluster size, enabling its application to medium-to-large solutes. The method is validated through the computation of Vibrational Circular Dichroism (VCD) spectra, which are highly sensitive to solute–solvent interactions. Test cases include aqueous L-alanine, aqueous dialanine, and (1S,2S)-trans-1-amino-2-indanol in DMSO. For L-alanine and trans-1-amino-2-indanol, computed spectra closely match experiment, with root-mean-square-deviation (RMSD) values of 10.3 and 8.0, respectively, consistent with previous benchmarks. For aqueous dialanine, the main spectral features were reproduced, though discrepancies in the fine structure remain, likely due to limitations in capturing subtle solvation effects. Overall, the refined EMCS protocol enables efficient and non-arbitrary sampling of solute–solvent clusters, offering a valuable tool for the structural analysis of solvation shells in complex molecular systems.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 17","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-06-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70144","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70144","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
The cluster-in-solvent approach, that is, the use of the quantum-mechanical calculation of a spectral observable on a significant number of solute–solvent clusters extracted from semi-classical simulations, is widely used in computational spectroscopy. However, identifying relevant coordinates for cluster selection remains a challenge. We previously developed the Ellipsoid Method for Cluster-in-Solvent (EMCS), an automated strategy for unbiased identification and statistical weighting of clusters. Yet, for larger solutes, EMCS can yield overly large solvent clusters that hinder conformational analysis. Here, we introduce a simple extension of EMCS that reduces cluster size, enabling its application to medium-to-large solutes. The method is validated through the computation of Vibrational Circular Dichroism (VCD) spectra, which are highly sensitive to solute–solvent interactions. Test cases include aqueous L-alanine, aqueous dialanine, and (1S,2S)-trans-1-amino-2-indanol in DMSO. For L-alanine and trans-1-amino-2-indanol, computed spectra closely match experiment, with root-mean-square-deviation (RMSD) values of 10.3 and 8.0, respectively, consistent with previous benchmarks. For aqueous dialanine, the main spectral features were reproduced, though discrepancies in the fine structure remain, likely due to limitations in capturing subtle solvation effects. Overall, the refined EMCS protocol enables efficient and non-arbitrary sampling of solute–solvent clusters, offering a valuable tool for the structural analysis of solvation shells in complex molecular systems.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


