Tatiana M Shamorkina, Laura Pérez Pañeda, Tereza Kadavá, Douwe Schulte, Patrick Pribil, Sibylle Heidelberger, Allison Michele Narlock-Brand, Steven M Yannone, Joost Snijder, Albert J R Heck
{"title":"Deep Coverage and Extended Sequence Reads Obtained with a Single Archaeal Protease Expedite <i>de novo</i> Protein Sequencing by Mass Spectrometry.","authors":"Tatiana M Shamorkina, Laura Pérez Pañeda, Tereza Kadavá, Douwe Schulte, Patrick Pribil, Sibylle Heidelberger, Allison Michele Narlock-Brand, Steven M Yannone, Joost Snijder, Albert J R Heck","doi":"10.1101/2025.05.26.656138","DOIUrl":null,"url":null,"abstract":"<p><p>The ability to sequence proteins without reliance on a genomic template defines a critical frontier in modern proteomics. This approach, known as <i>de novo</i> protein sequencing, is essential for applications such as antibody sequencing, microbiome proteomics, and antigen discovery, which require accurate reconstruction of peptide and protein sequences. While trypsin remains the gold-standard protease in proteomics, its restricted cleavage specificity limits peptide diversity. This constraint is especially problematic in antibody sequencing, where the functionally critical regions often lack canonical tryptic sites. As a result, exclusively trypsin-based approaches yield sparse reads, leading to sequence gaps. Multi-protease and hybrid-fragmentation strategies can improve the sequence coverage, but they add complexity, compromise scalability and reproducibility. Here, we explore two HyperThermoacidic Archaeal (HTA)-proteases as single-enzyme solutions for <i>de novo</i> antibody sequencing. Each HTA-protease generated about five times more unique peptide reads than trypsin or chymotrypsin, providing high redundancy across all CDRs. Combined with EAciD fragmentation on a ZenoTOF 7600 system, this approach enabled complete, unambiguous antibody sequencing. <i>De novo</i> analysis using PEAKS/DeepNovo and Stitch showed up to fourfold higher alignment scores and reduced the sequence errors within the HTA-generated data. Additionally, the HTA-EAciD approach offers short digestion times, eliminates extensive cleanup, and enables analysis in a single LC-MS/MS run. This streamlined, single-protease approach delivers therefore performance comparable to multi-enzyme workflows, offering a scalable and efficient strategy for <i>de novo</i> protein sequencing across diverse applications.</p>","PeriodicalId":519960,"journal":{"name":"bioRxiv : the preprint server for biology","volume":" ","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2025-05-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12154832/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"bioRxiv : the preprint server for biology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1101/2025.05.26.656138","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
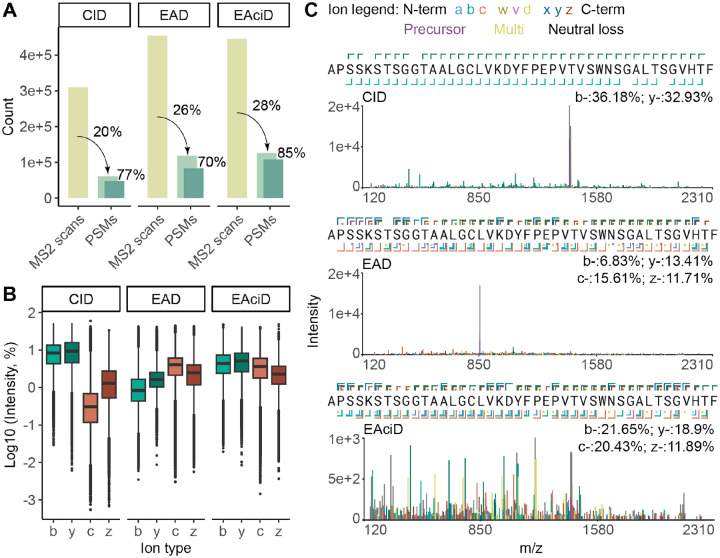
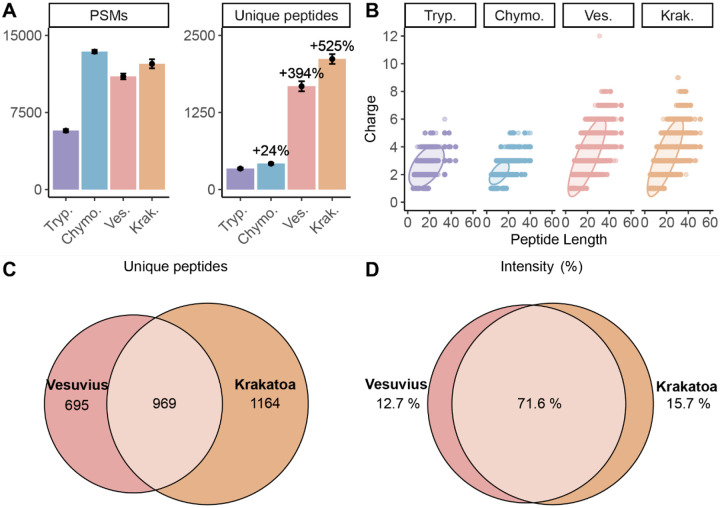
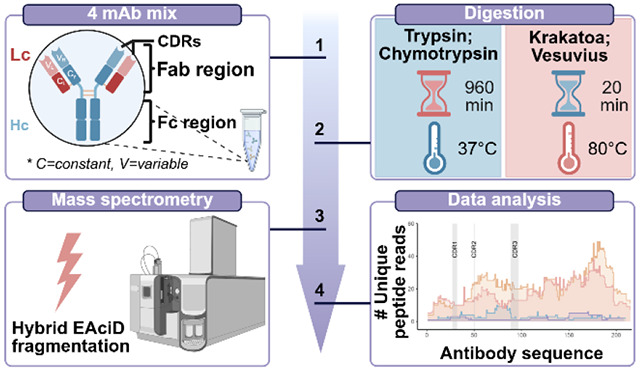
The ability to sequence proteins without reliance on a genomic template defines a critical frontier in modern proteomics. This approach, known as de novo protein sequencing, is essential for applications such as antibody sequencing, microbiome proteomics, and antigen discovery, which require accurate reconstruction of peptide and protein sequences. While trypsin remains the gold-standard protease in proteomics, its restricted cleavage specificity limits peptide diversity. This constraint is especially problematic in antibody sequencing, where the functionally critical regions often lack canonical tryptic sites. As a result, exclusively trypsin-based approaches yield sparse reads, leading to sequence gaps. Multi-protease and hybrid-fragmentation strategies can improve the sequence coverage, but they add complexity, compromise scalability and reproducibility. Here, we explore two HyperThermoacidic Archaeal (HTA)-proteases as single-enzyme solutions for de novo antibody sequencing. Each HTA-protease generated about five times more unique peptide reads than trypsin or chymotrypsin, providing high redundancy across all CDRs. Combined with EAciD fragmentation on a ZenoTOF 7600 system, this approach enabled complete, unambiguous antibody sequencing. De novo analysis using PEAKS/DeepNovo and Stitch showed up to fourfold higher alignment scores and reduced the sequence errors within the HTA-generated data. Additionally, the HTA-EAciD approach offers short digestion times, eliminates extensive cleanup, and enables analysis in a single LC-MS/MS run. This streamlined, single-protease approach delivers therefore performance comparable to multi-enzyme workflows, offering a scalable and efficient strategy for de novo protein sequencing across diverse applications.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


