Matteo Orlandi, Marina Macchiagodena, Piero Procacci, Fabrizio Carta, Claudiu T. Supuran, Marco Pagliai
{"title":"Development of a New AMBER Force Field for Cysteine and Histidine Cadmium-Binding Proteins and Its Validation Through QM/MM MD Simulations","authors":"Matteo Orlandi, Marina Macchiagodena, Piero Procacci, Fabrizio Carta, Claudiu T. Supuran, Marco Pagliai","doi":"10.1002/jcc.70154","DOIUrl":null,"url":null,"abstract":"<p>We developed and validated a novel force field in the context of the AMBER parameterization for the simulation of cadmium(II)-binding proteins. The proposed force field takes into account the polarization effect produced by the central ion on its surroundings. The new polarized atomic charges for cysteine and histidine residues were derived based on the available structures of cadmium-bearing proteins using QM calculations and QM/MM simulations. The developed force field was validated by performing molecular dynamics simulations on several cadmium(II)-binding proteins. Our model preserves the tetra-coordination of the metal site with remarkable stability, yielding mean distances between <span></span><math>\n <semantics>\n <mrow>\n <msup>\n <mi>Cd</mi>\n <mrow>\n <mn>2</mn>\n <mo>+</mo>\n </mrow>\n </msup>\n </mrow>\n <annotation>$$ {\\mathrm{Cd}}^{2+} $$</annotation>\n </semantics></math> ion and S or N atoms of the binding residues in close agreement with experimental data.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 16","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2025-06-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.70154","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.70154","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
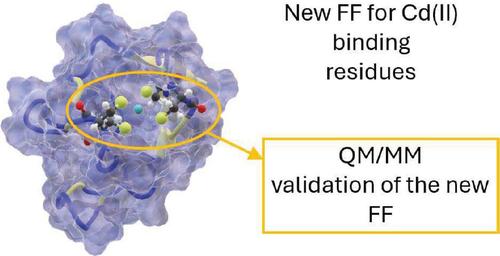
We developed and validated a novel force field in the context of the AMBER parameterization for the simulation of cadmium(II)-binding proteins. The proposed force field takes into account the polarization effect produced by the central ion on its surroundings. The new polarized atomic charges for cysteine and histidine residues were derived based on the available structures of cadmium-bearing proteins using QM calculations and QM/MM simulations. The developed force field was validated by performing molecular dynamics simulations on several cadmium(II)-binding proteins. Our model preserves the tetra-coordination of the metal site with remarkable stability, yielding mean distances between ion and S or N atoms of the binding residues in close agreement with experimental data.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


