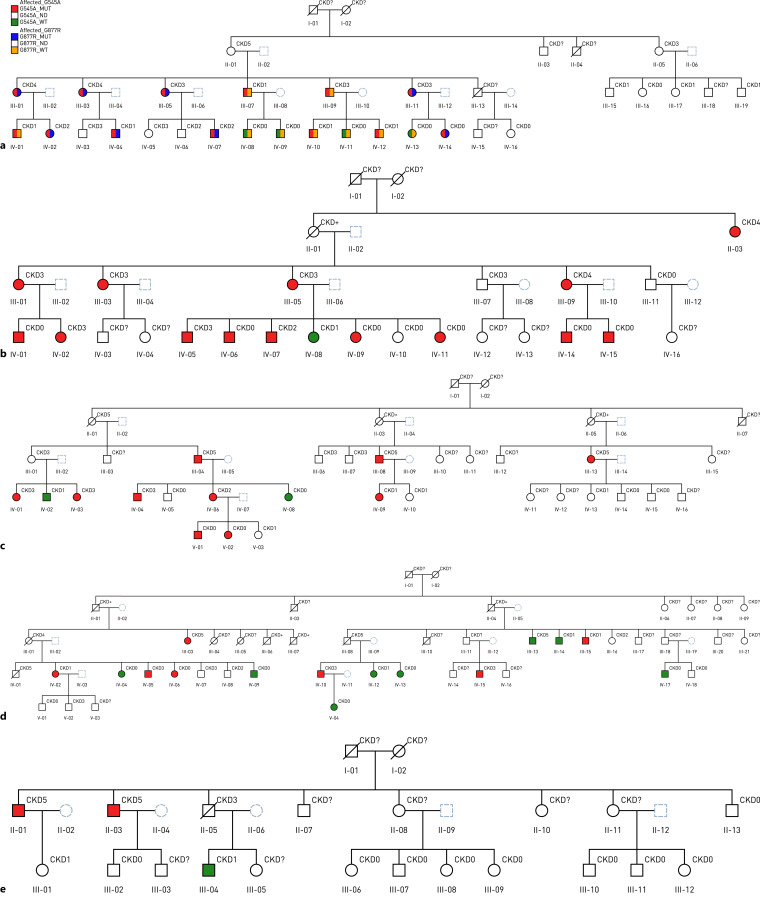
Fezile Ozdemir, D Deren Oygar, Ahmet Behlul, Salahi Ataç, Simge Bardak, Meral Yükseliş, Gregory Papagregoriou, Apostolos Malatras, Daniel P Gale, Guy H Neild, Constantinos Deltas, Cemal Gurkan
{"title":"Familial Kidney Disease Phenocopying Hypertensive Nephropathy.","authors":"Fezile Ozdemir, D Deren Oygar, Ahmet Behlul, Salahi Ataç, Simge Bardak, Meral Yükseliş, Gregory Papagregoriou, Apostolos Malatras, Daniel P Gale, Guy H Neild, Constantinos Deltas, Cemal Gurkan","doi":"10.1159/000546094","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Familial kidney disease is common in Cyprus and previous studies have found that the majority of families have mutations in Alport syndrome genes COL4A3/4/5. We have collected data from over 50 Turkish Cypriot families in whom kidney disease appears to follow an autosomal dominant pattern, and looked for pathological variants in these genes.</p><p><strong>Methods: </strong>Probands from 55 families underwent massive parallel DNA sequencing using a glomerular gene panel for familial hematuria, and whole-exome sequencing (WES) was also performed in 22 of them. Clinical records were reviewed.</p><p><strong>Results: </strong>Likely pathogenic variants were identified in 7 of the 55 families (COL4A3 [3], COL4A4 [2], and COL4A5 [2]), leaving 48 unsolved families. Among the latter a common missense variant of uncertain significance (COL4A4:p.G545A), was present in 5 families (9.1%). In contrast to families with a pathogenic variant in COL4A3/4 and a clear glomerular phenotype the 5 families (54 patients with clinical and genetic data), manifested near dominant susceptibility with incomplete penetrance, presenting with hypertension, variable and intermittent microscopic hematuria, and minimal proteinuria, <1 g/day until the estimated glomerular filtration rate (eGFR) fell below 30 mL/min, after which it increased in some individuals. Of those over age 50, 20% had reached end-stage by median age of 66 (48-80) years.</p><p><strong>Conclusions: </strong>We describe a kidney disease with mild hypertension that is more characteristic of a tubulointerstitial disease and phenocopies hypertensive nephropathy. While the variant COL4A4:p.G545A is not responsible for a Mendelian CKD phenotype, it appears to increases the susceptibility, acting as a hypomorphic variant contributing to Alport spectrum nephropathy. Early detection and treatment with ACE inhibitors should prolong kidney survival to an age where hemodialysis is avoided.</p>","PeriodicalId":73177,"journal":{"name":"Glomerular diseases","volume":"5 1","pages":"233-242"},"PeriodicalIF":0.0000,"publicationDate":"2025-04-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12148318/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Glomerular diseases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000546094","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Familial kidney disease is common in Cyprus and previous studies have found that the majority of families have mutations in Alport syndrome genes COL4A3/4/5. We have collected data from over 50 Turkish Cypriot families in whom kidney disease appears to follow an autosomal dominant pattern, and looked for pathological variants in these genes.
Methods: Probands from 55 families underwent massive parallel DNA sequencing using a glomerular gene panel for familial hematuria, and whole-exome sequencing (WES) was also performed in 22 of them. Clinical records were reviewed.
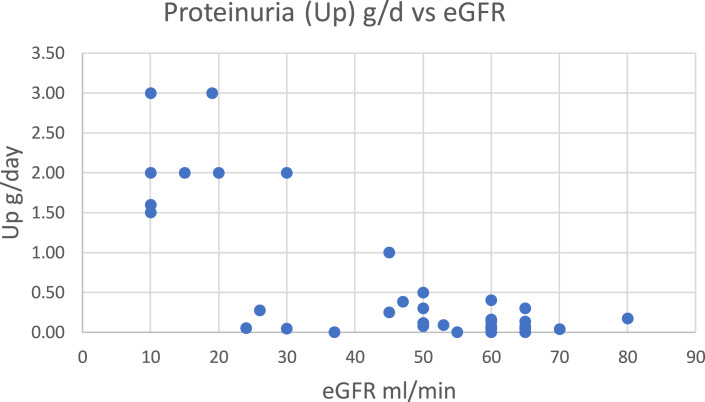
Results: Likely pathogenic variants were identified in 7 of the 55 families (COL4A3 [3], COL4A4 [2], and COL4A5 [2]), leaving 48 unsolved families. Among the latter a common missense variant of uncertain significance (COL4A4:p.G545A), was present in 5 families (9.1%). In contrast to families with a pathogenic variant in COL4A3/4 and a clear glomerular phenotype the 5 families (54 patients with clinical and genetic data), manifested near dominant susceptibility with incomplete penetrance, presenting with hypertension, variable and intermittent microscopic hematuria, and minimal proteinuria, <1 g/day until the estimated glomerular filtration rate (eGFR) fell below 30 mL/min, after which it increased in some individuals. Of those over age 50, 20% had reached end-stage by median age of 66 (48-80) years.
Conclusions: We describe a kidney disease with mild hypertension that is more characteristic of a tubulointerstitial disease and phenocopies hypertensive nephropathy. While the variant COL4A4:p.G545A is not responsible for a Mendelian CKD phenotype, it appears to increases the susceptibility, acting as a hypomorphic variant contributing to Alport spectrum nephropathy. Early detection and treatment with ACE inhibitors should prolong kidney survival to an age where hemodialysis is avoided.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


