{"title":"Reconstructing rearrangement phylogenies of natural genomes.","authors":"Leonard Bohnenkämper, Jens Stoye, Daniel Doerr","doi":"10.1186/s13015-025-00279-5","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>We study the classical problem of inferring ancestral genomes from a set of extant genomes under a given phylogeny, known as the Small Parsimony Problem (SPP). Genomes are represented as sequences of oriented markers, organized in one or more linear or circular chromosomes. Any marker may appear in several copies, without restriction on orientation or genomic location, known as the natural genomes model. Evolutionary events along the branches of the phylogeny encompass large scale rearrangements, including segmental inversions, translocations, gain and loss (DCJ-indel model). Even under simpler rearrangement models, such as the classical breakpoint model without duplicates, the SPP is computationally intractable. Nevertheless, the SPP for natural genomes under the DCJ-indel model has been studied recently, with limited success.</p><p><strong>Methods: </strong>Building on prior work, we present a highly optimized ILP that is able to solve the SPP for sufficiently small phylogenies and gene families. A notable improvement w.r.t. the previous result is an optimized way of handling both circular and linear chromosomes. This is especially relevant to the SPP, since the chromosomal structure of ancestral genomes is unknown and the solution space for this chromosomal structure is typically large.</p><p><strong>Results: </strong>We benchmark our method on simulated and real data. On simulated phylogenies we observe a considerable performance improvement on problems that include linear chromosomes. And even when the ground truth contains only one circular chromosome per genome, our method outperforms its predecessor due to its optimized handling of the solution space. The practical advantage becomes also visible in an analysis of seven Anopheles taxa.</p>","PeriodicalId":50823,"journal":{"name":"Algorithms for Molecular Biology","volume":"20 1","pages":"10"},"PeriodicalIF":1.7000,"publicationDate":"2025-06-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12144824/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Algorithms for Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1186/s13015-025-00279-5","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: We study the classical problem of inferring ancestral genomes from a set of extant genomes under a given phylogeny, known as the Small Parsimony Problem (SPP). Genomes are represented as sequences of oriented markers, organized in one or more linear or circular chromosomes. Any marker may appear in several copies, without restriction on orientation or genomic location, known as the natural genomes model. Evolutionary events along the branches of the phylogeny encompass large scale rearrangements, including segmental inversions, translocations, gain and loss (DCJ-indel model). Even under simpler rearrangement models, such as the classical breakpoint model without duplicates, the SPP is computationally intractable. Nevertheless, the SPP for natural genomes under the DCJ-indel model has been studied recently, with limited success.
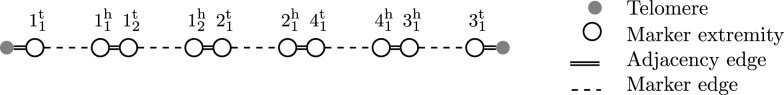
Methods: Building on prior work, we present a highly optimized ILP that is able to solve the SPP for sufficiently small phylogenies and gene families. A notable improvement w.r.t. the previous result is an optimized way of handling both circular and linear chromosomes. This is especially relevant to the SPP, since the chromosomal structure of ancestral genomes is unknown and the solution space for this chromosomal structure is typically large.
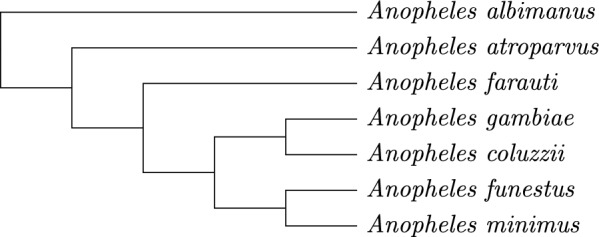
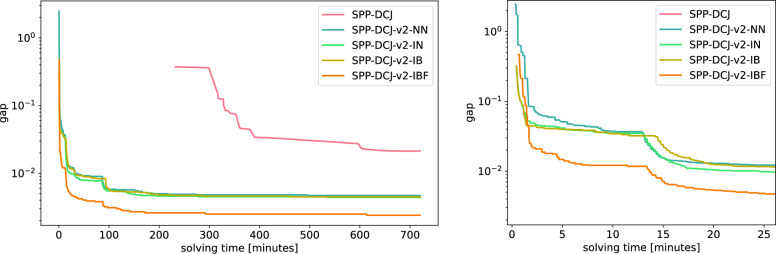
Results: We benchmark our method on simulated and real data. On simulated phylogenies we observe a considerable performance improvement on problems that include linear chromosomes. And even when the ground truth contains only one circular chromosome per genome, our method outperforms its predecessor due to its optimized handling of the solution space. The practical advantage becomes also visible in an analysis of seven Anopheles taxa.
期刊介绍:
Algorithms for Molecular Biology publishes articles on novel algorithms for biological sequence and structure analysis, phylogeny reconstruction, and combinatorial algorithms and machine learning.
Areas of interest include but are not limited to: algorithms for RNA and protein structure analysis, gene prediction and genome analysis, comparative sequence analysis and alignment, phylogeny, gene expression, machine learning, and combinatorial algorithms.
Where appropriate, manuscripts should describe applications to real-world data. However, pure algorithm papers are also welcome if future applications to biological data are to be expected, or if they address complexity or approximation issues of novel computational problems in molecular biology. Articles about novel software tools will be considered for publication if they contain some algorithmically interesting aspects.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


