Assessing simulation-based supervised machine learning for demographic parameter inference from genomic data
IF 3.9
2区 生物学
Q2 ECOLOGY
引用次数: 0
Abstract
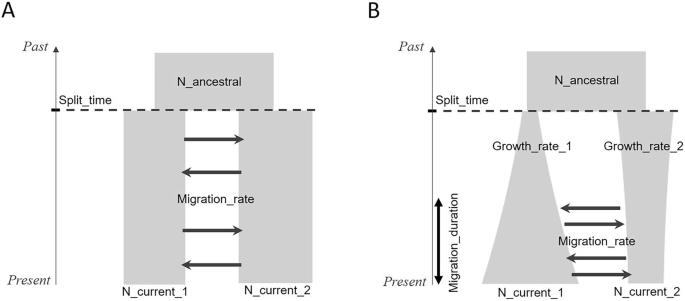
The ever-increasing availability of high-throughput DNA sequences and the development of numerous computational methods have led to considerable advances in our understanding of the evolutionary and demographic history of populations. Several demographic inference methods have been developed to take advantage of these massive genomic data. Simulation-based approaches, such as approximate Bayesian computation (ABC), have proved particularly efficient for complex demographic models. However, taking full advantage of the comprehensive information contained in massive genomic data remains a challenge for demographic inference methods, which generally rely on partial information from these data. Using advanced computational methods, such as machine learning, is valuable for efficiently integrating more comprehensive information. Here, we showed how simulation-based supervised machine learning methods applied to an extensive range of summary statistics are effective in inferring demographic parameters for connected populations. We compared three machine learning (ML) methods: a neural network, the multilayer perceptron (MLP), and two ensemble methods, random forest (RF) and the gradient boosting system XGBoost (XGB), to infer demographic parameters from genomic data under a standard isolation with migration model and a secondary contact model with varying population sizes. We showed that MLP outperformed the other two methods and that, on the basis of permutation feature importance, its predictions involved a larger combination of summary statistics. Moreover, they outperformed all three tested ABC algorithms. Finally, we demonstrated how a method called SHAP, from the field of explainable artificial intelligence, can be used to shed light on the contribution of summary statistics within the ML models.
评估基于模拟的监督机器学习对基因组数据的人口统计参数推断。
高通量DNA序列的不断增加的可用性和许多计算方法的发展导致我们对种群进化和人口历史的理解取得了相当大的进步。为了利用这些海量的基因组数据,已经开发了几种人口统计学推断方法。基于模拟的方法,如近似贝叶斯计算(ABC),已被证明对复杂的人口统计模型特别有效。然而,充分利用海量基因组数据中包含的综合信息仍然是人口统计推断方法的一个挑战,这些方法通常依赖于这些数据中的部分信息。使用先进的计算方法,如机器学习,对于有效地集成更全面的信息是有价值的。在这里,我们展示了基于模拟的监督机器学习方法如何应用于广泛的汇总统计,在推断连接人群的人口统计参数方面是有效的。我们比较了三种机器学习(ML)方法:一种神经网络,多层感知器(MLP),以及两种集成方法,随机森林(RF)和梯度增强系统XGBoost (XGB),在标准隔离迁移模型和不同人口规模的二次接触模型下从基因组数据推断人口统计学参数。我们表明,MLP优于其他两种方法,并且在排列特征重要性的基础上,其预测涉及更大的汇总统计组合。此外,它们的表现优于所有三种经过测试的ABC算法。最后,我们演示了如何使用来自可解释人工智能领域的SHAP方法来阐明ML模型中汇总统计的贡献。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Heredity
生物-进化生物学
CiteScore
7.50
自引率
2.60%
发文量
84
审稿时长
4-8 weeks
期刊介绍:
Heredity is the official journal of the Genetics Society. It covers a broad range of topics within the field of genetics and therefore papers must address conceptual or applied issues of interest to the journal''s wide readership
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


