Xia Zhou, Shengyou Su, Shenghua Li, ZuFang Yi, Liling Feng, Junyi Chen, Binglin Fan
{"title":"Type I Sialidosis in a Chinese family: a case report and literature review.","authors":"Xia Zhou, Shengyou Su, Shenghua Li, ZuFang Yi, Liling Feng, Junyi Chen, Binglin Fan","doi":"10.1186/s42494-025-00225-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Sialidosis is an autosomal recessive hereditary disease characterized by the mutation of neuraminidase-1 (NEU1) gene, resulting in decreased activity of α-N-acetylneuraminidase. This leads to metabolic abnormalities in various organs. Sialidosis is classified into two distinct clinical phenotypes, type I and type II, based on the age of onset and severity of clinical manifestations.</p><p><strong>Case presentation: </strong>Here, we report a case involving a patient and his two sisters, all of whom showed seizures and ataxia during adolescence, with progressively worsening symptoms. Prior to admission, none of the patients had received a systemic diagnosis or treatment. The whole exome sequencing identified a homozygous NEU1 mutation (NM_000434.3:c.544A > G [p.Ser182Gly]) in all three siblings. Their parents and children, who were asymptomatic, were found to be heterozygous carriers. The three patients were ultimately diagnosed with type I sialidosis and treated with antiseizure medications, but they continued to experience recurrent seizures.</p><p><strong>Conclusions: </strong>This case report enhances our understanding of sialidosis, particularly in patients presenting with seizures and ataxia. Furthermore, the gene sequencing is a crucial tool for confirming the diagnosis of sialidosis and provides a valuable approach for genetic counseling in affected families.</p>","PeriodicalId":33628,"journal":{"name":"Acta Epileptologica","volume":"7 1","pages":"34"},"PeriodicalIF":1.2000,"publicationDate":"2025-06-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12135267/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Epileptologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42494-025-00225-3","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Sialidosis is an autosomal recessive hereditary disease characterized by the mutation of neuraminidase-1 (NEU1) gene, resulting in decreased activity of α-N-acetylneuraminidase. This leads to metabolic abnormalities in various organs. Sialidosis is classified into two distinct clinical phenotypes, type I and type II, based on the age of onset and severity of clinical manifestations.
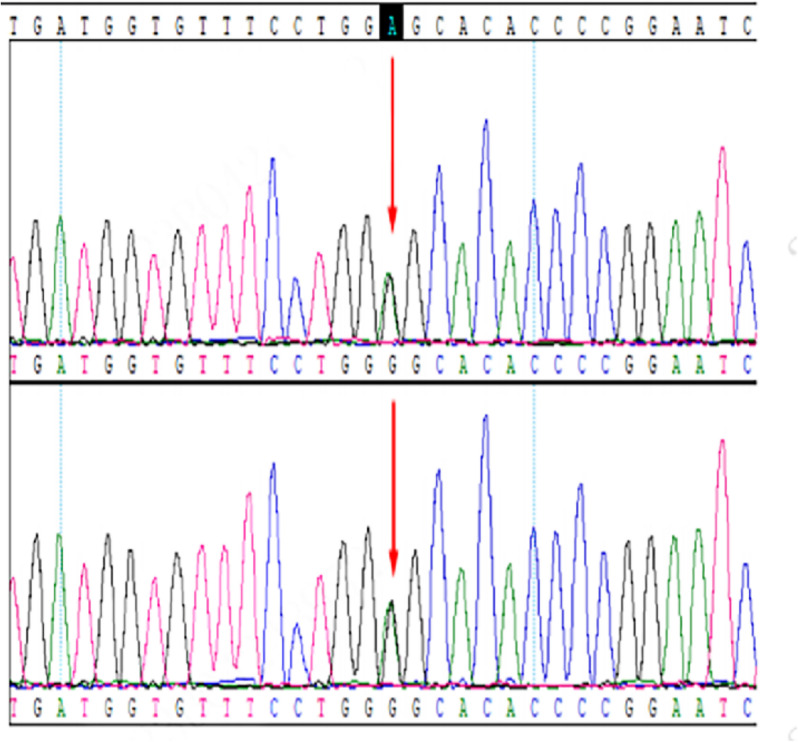
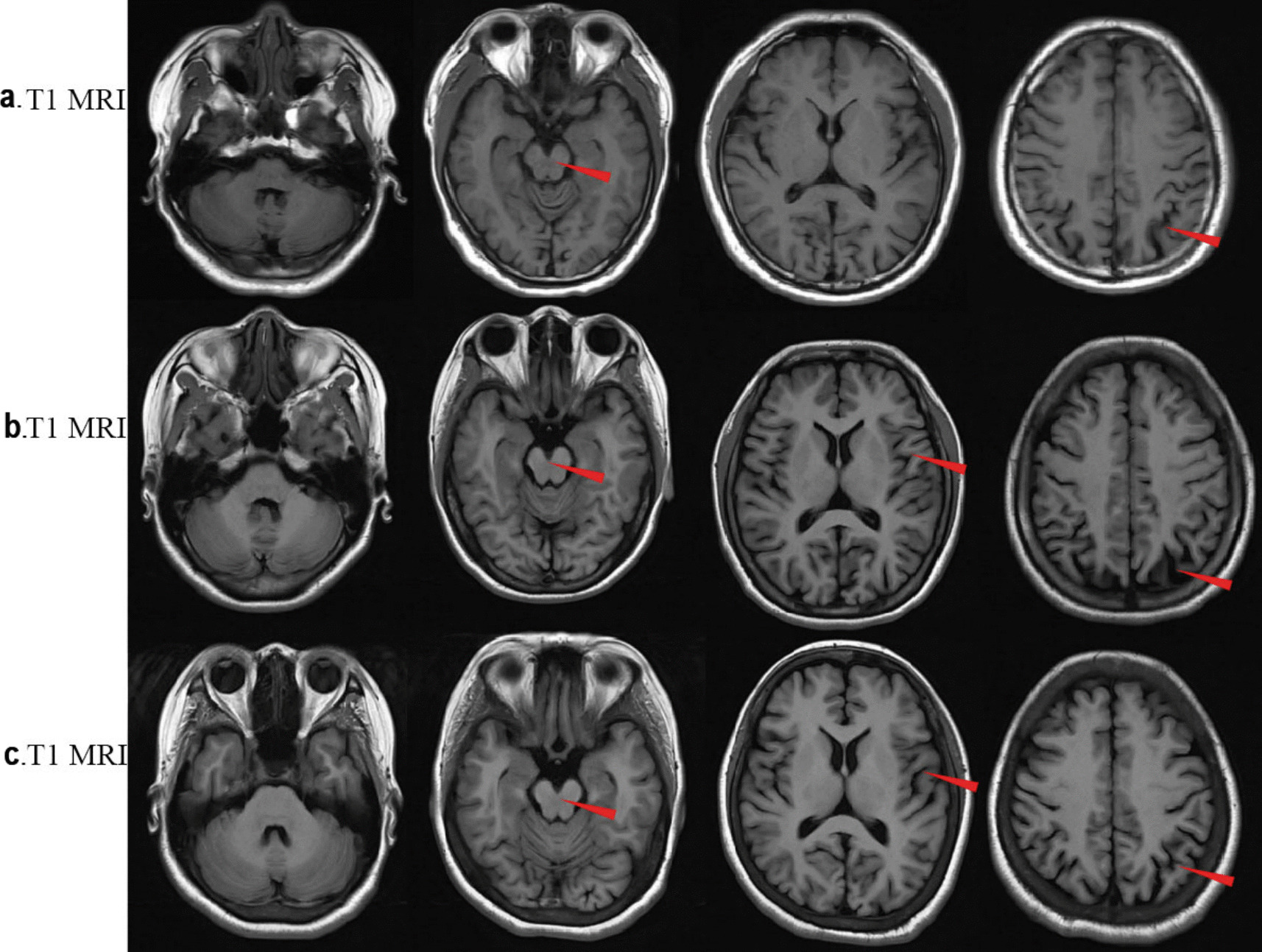
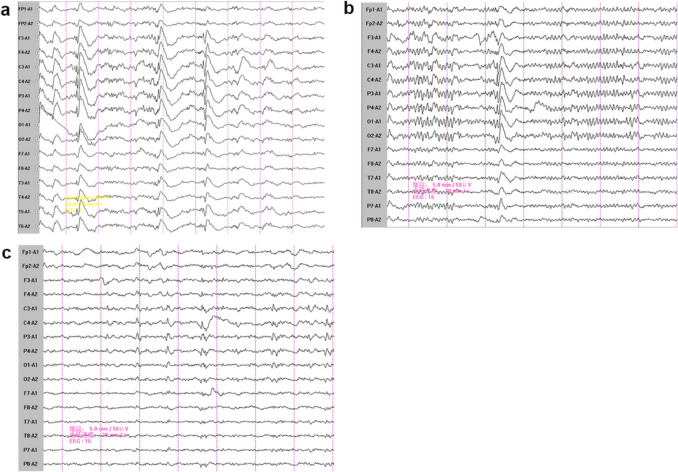
Case presentation: Here, we report a case involving a patient and his two sisters, all of whom showed seizures and ataxia during adolescence, with progressively worsening symptoms. Prior to admission, none of the patients had received a systemic diagnosis or treatment. The whole exome sequencing identified a homozygous NEU1 mutation (NM_000434.3:c.544A > G [p.Ser182Gly]) in all three siblings. Their parents and children, who were asymptomatic, were found to be heterozygous carriers. The three patients were ultimately diagnosed with type I sialidosis and treated with antiseizure medications, but they continued to experience recurrent seizures.
Conclusions: This case report enhances our understanding of sialidosis, particularly in patients presenting with seizures and ataxia. Furthermore, the gene sequencing is a crucial tool for confirming the diagnosis of sialidosis and provides a valuable approach for genetic counseling in affected families.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


