Isoform switch of T-cell factor7L2 during mouse heart development
IF 2.2
Journal of molecular and cellular cardiology plus
Pub Date : 2025-06-01
DOI:10.1016/j.jmccpl.2025.100458
引用次数: 0
Abstract
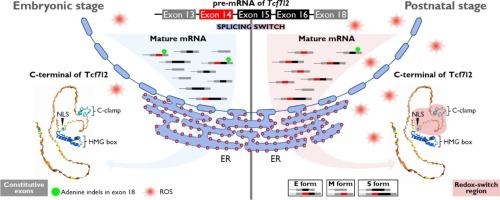
Canonical WNT signaling plays critical, often opposing roles in heart development and disease, but its context-dependent mechanisms remain unclear. We hypothesized that alternative splicing of Tcf7l2, a key nuclear partner of β-catenin, contributes to WNT signaling specificity in the heart. To investigate this, we cloned and sequenced 53 Tcf7l2 transcripts in ventricular tissues from embryonic day 17.5 (E17.5, 24/53) and postnatal day 8 (P8, 29/53) mice, identifying 32 distinct isoforms. Among 18 potential exons, exons 6 and 17 were absent, and over 80 % of transcripts lacked exon 4. Alternative splicing was prominent in the C-terminal exons (14, 15, and 16), with exon 14 inclusion significantly higher in P8 hearts (64.3 %) than E17.5 hearts (34.8 %). Variations in exon 15 and 16 combinations, along with reading frame shifts caused by the adenine insertion and deletion (indel) near the beginning of exon 18, affected C-terminal structures, altering the presence of the E-tail, C-clamp, and CtBP-binding motifs. Notably, exon 14 insertion introduced a redox-switch domain spanning the NLS and C-clamp regions in E and S isoforms, while adenine indels altered isoform lengths, driving transitions between E, S, and M isoforms. RT-PCR validation across multiple developmental stages confirmed these splicing patterns. Our findings suggest that a postnatal redox-sensitive isoform switch in Tcf7l2 modulates WNT signaling, potentially influencing cardiomyocyte maturation during the transition from proliferation to hypertrophy.
小鼠心脏发育过程中t细胞因子7l2的异构体开关
典型的WNT信号在心脏发育和疾病中起着关键的,通常是相反的作用,但其上下文依赖的机制尚不清楚。我们假设β-catenin的关键核伴侣Tcf7l2的选择性剪接有助于心脏中WNT信号的特异性。为了研究这一点,我们克隆并测序了胚胎17.5天(E17.5, 24/53)和出生后第8天(P8, 29/53)小鼠心室组织中的53个Tcf7l2转录本,鉴定了32个不同的亚型。在18个潜在外显子中,外显子6和17缺失,超过80%的转录本缺乏外显子4。选择性剪接在c端外显子(14、15和16)中很突出,P8心脏的外显子14包含率(64.3%)显著高于E17.5心脏(34.8%)。外显子15和16组合的变化,以及由靠近外显子18开头的腺嘌呤插入和缺失(indel)引起的阅读框移位,影响了c端结构,改变了E-tail、C-clamp和ctbp结合基序的存在。值得注意的是,外显子14的插入在E和S异构体中引入了一个跨越NLS和C-clamp区域的氧化还原开关结构域,而腺嘌呤插入改变了异构体的长度,驱动了E、S和M异构体之间的转换。跨多个发育阶段的RT-PCR验证证实了这些剪接模式。我们的研究结果表明,Tcf7l2的出生后氧化还原敏感异构体开关调节WNT信号,可能影响心肌细胞从增殖到肥大转变过程中的成熟。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular and cellular cardiology plus
Cardiology and Cardiovascular Medicine
自引率
0.00%
发文量
0
审稿时长
31 days
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


