Rawan Shraim, Caroline Diorio, Scott W Canna, Erin Macdonald-Dunlop, Hamid Bassiri, Zachary Martinez, Anders Mälarstig, Afrouz Abbaspour, David T Teachey, Robert B Lindell, Edward M Behrens
{"title":"A Method for Comparing Proteins Measured in Serum and Plasma by Olink Proximity Extension Assay.","authors":"Rawan Shraim, Caroline Diorio, Scott W Canna, Erin Macdonald-Dunlop, Hamid Bassiri, Zachary Martinez, Anders Mälarstig, Afrouz Abbaspour, David T Teachey, Robert B Lindell, Edward M Behrens","doi":"10.1016/j.mcpro.2025.101000","DOIUrl":null,"url":null,"abstract":"<p><p>Accurate measurement of secreted proteins in serum and plasma is essential for understanding mechanisms and developing reliable biomarkers. Recent technological advancements, such as proximity extension assay (PEA), have enabled high-throughput multiplex protein analyses from small sample volumes in either serum or plasma. Despite the increasing use of PEA-based proteomics and the generation of extensive datasets, integrated data from these two mediums remains challenging due to inherent differences in sample processing. To address this issue, we developed and validated protein-specific transformation factors using linear modeling to normalize protein measurements between serum and plasma proteins quantified using Olink. Our analysis surveyed 1463 proteins across matched serum and plasma samples, identifying 686 transformation factors. The transformation factors were further validated using independent datasets generated from patients with different disease phenotypes and ages, and 551 of the models and transformation factors were reproducible. These transformation factors provide a valuable resource for normalizing PEA-based proteomic data across serum and plasma, ultimately enhancing the capacity for collaborative analyses and facilitating comprehensive insights across diverse disease phenotypes.</p>","PeriodicalId":18712,"journal":{"name":"Molecular & Cellular Proteomics","volume":" ","pages":"101000"},"PeriodicalIF":5.5000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12418474/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular & Cellular Proteomics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1016/j.mcpro.2025.101000","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/27 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
引用次数: 0
Abstract
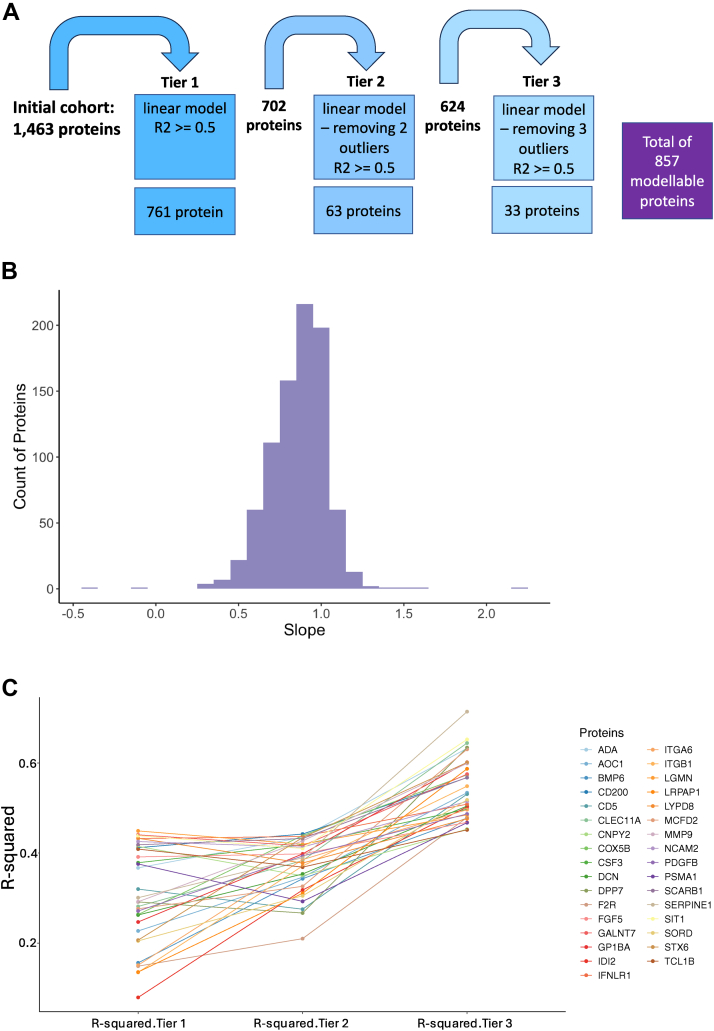
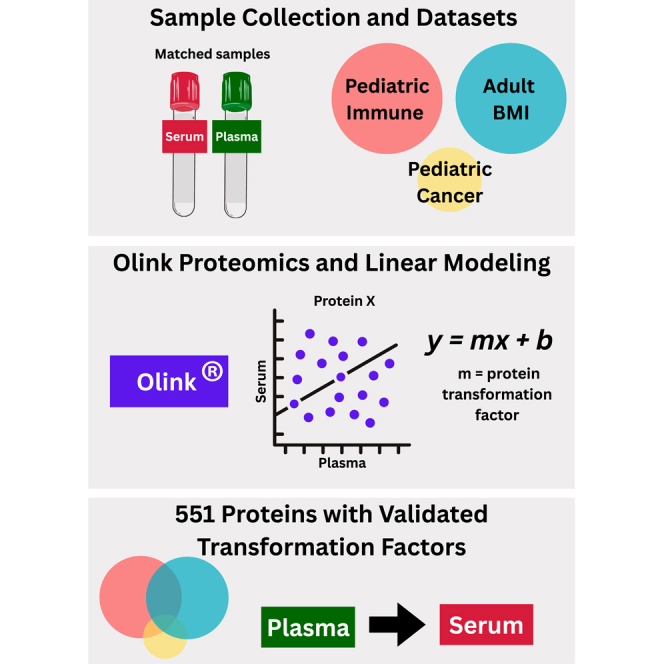
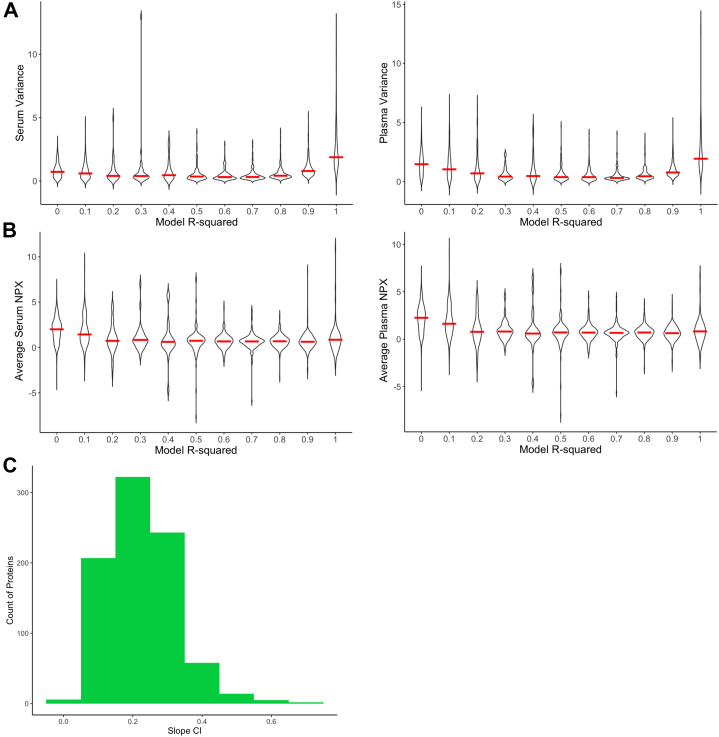
Accurate measurement of secreted proteins in serum and plasma is essential for understanding mechanisms and developing reliable biomarkers. Recent technological advancements, such as proximity extension assay (PEA), have enabled high-throughput multiplex protein analyses from small sample volumes in either serum or plasma. Despite the increasing use of PEA-based proteomics and the generation of extensive datasets, integrated data from these two mediums remains challenging due to inherent differences in sample processing. To address this issue, we developed and validated protein-specific transformation factors using linear modeling to normalize protein measurements between serum and plasma proteins quantified using Olink. Our analysis surveyed 1463 proteins across matched serum and plasma samples, identifying 686 transformation factors. The transformation factors were further validated using independent datasets generated from patients with different disease phenotypes and ages, and 551 of the models and transformation factors were reproducible. These transformation factors provide a valuable resource for normalizing PEA-based proteomic data across serum and plasma, ultimately enhancing the capacity for collaborative analyses and facilitating comprehensive insights across diverse disease phenotypes.
期刊介绍:
The mission of MCP is to foster the development and applications of proteomics in both basic and translational research. MCP will publish manuscripts that report significant new biological or clinical discoveries underpinned by proteomic observations across all kingdoms of life. Manuscripts must define the biological roles played by the proteins investigated or their mechanisms of action.
The journal also emphasizes articles that describe innovative new computational methods and technological advancements that will enable future discoveries. Manuscripts describing such approaches do not have to include a solution to a biological problem, but must demonstrate that the technology works as described, is reproducible and is appropriate to uncover yet unknown protein/proteome function or properties using relevant model systems or publicly available data.
Scope:
-Fundamental studies in biology, including integrative "omics" studies, that provide mechanistic insights
-Novel experimental and computational technologies
-Proteogenomic data integration and analysis that enable greater understanding of physiology and disease processes
-Pathway and network analyses of signaling that focus on the roles of post-translational modifications
-Studies of proteome dynamics and quality controls, and their roles in disease
-Studies of evolutionary processes effecting proteome dynamics, quality and regulation
-Chemical proteomics, including mechanisms of drug action
-Proteomics of the immune system and antigen presentation/recognition
-Microbiome proteomics, host-microbe and host-pathogen interactions, and their roles in health and disease
-Clinical and translational studies of human diseases
-Metabolomics to understand functional connections between genes, proteins and phenotypes

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


