A novel c.116 - 117 del variant in Unverricht-Lundborg disease: first ULD report in large Chinese population and review of the pathogenetic variants in CSTB gene.
Pu Miao, Yao Ding, Zhidong Cen, Yulan Chen, Wei Luo, Baorong Zhang, Zhiying Wu, Meiping Ding, Shuang Wang
{"title":"A novel c.116 - 117 del variant in Unverricht-Lundborg disease: first ULD report in large Chinese population and review of the pathogenetic variants in CSTB gene.","authors":"Pu Miao, Yao Ding, Zhidong Cen, Yulan Chen, Wei Luo, Baorong Zhang, Zhiying Wu, Meiping Ding, Shuang Wang","doi":"10.1186/s42494-025-00216-4","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Unverricht-Lundborg disease (ULD) is a rare autosomal recessive neurodegenerative disorder, often caused by biallelic promoter expansions of CSTB gene or, more rarely by point/indel variants. The best-known area for ULD are the shores of the Baltic and Mediterranean Sea and few cases have been recorded from Asia.</p><p><strong>Case presentation: </strong>In this report, we present the first case of a Chinese patient with ULD. The patient was a 21-year-old female with normal cognitive function. She developed nocturnal bilateral tonic-clonic seizures (BTCS) at age 8, with subsequent onset of myoclonic jerks along with ataxia at age 12. Myoclonic jerks were triggered by flashing lights and during menstrual periods. EEG recording showed multifocal spikes and sharp-waves, predominantly in bilateral occipital regions. Genetic testing revealed heterozygous compound variants for a novel indel variant (c.116 - 117 delAG) and the repeat expansion of CSTB gene. The refractory BTCS and myoclonic jerks showed remarkable response to low-dose (2 mg/day) perampanel treatment. After 24 months of follow-up, the patient remained seizure-free, but her myoclonic jerks recurred, which could be reduced by increasing the dosage of perampanel.</p><p><strong>Conclusions: </strong>To the best of our knowledge, this is the first report of ULD in the large Chinese population. By comparison with homozygous promoter expansions, we found an earlier age of first symptom onset and more refractory BTCS of ULD patients with compound or homozygous point/indel variants.</p>","PeriodicalId":33628,"journal":{"name":"Acta Epileptologica","volume":"7 1","pages":"32"},"PeriodicalIF":1.2000,"publicationDate":"2025-05-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12121152/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Acta Epileptologica","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1186/s42494-025-00216-4","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"CLINICAL NEUROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
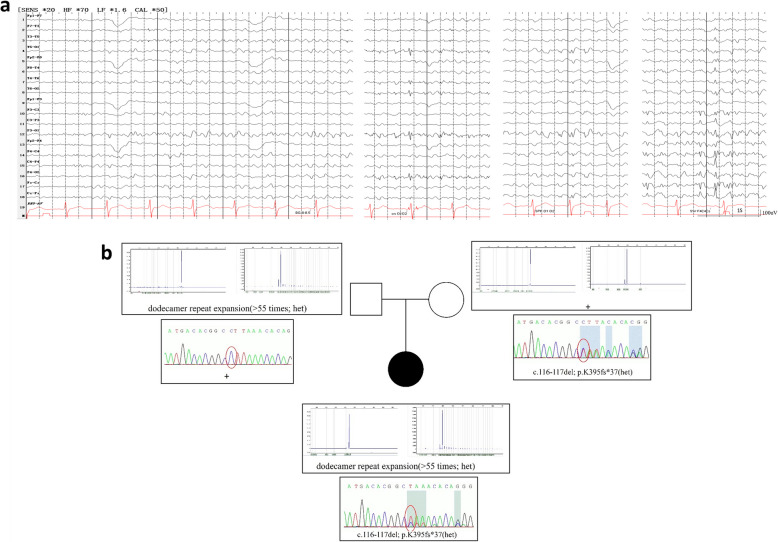
Background: Unverricht-Lundborg disease (ULD) is a rare autosomal recessive neurodegenerative disorder, often caused by biallelic promoter expansions of CSTB gene or, more rarely by point/indel variants. The best-known area for ULD are the shores of the Baltic and Mediterranean Sea and few cases have been recorded from Asia.
Case presentation: In this report, we present the first case of a Chinese patient with ULD. The patient was a 21-year-old female with normal cognitive function. She developed nocturnal bilateral tonic-clonic seizures (BTCS) at age 8, with subsequent onset of myoclonic jerks along with ataxia at age 12. Myoclonic jerks were triggered by flashing lights and during menstrual periods. EEG recording showed multifocal spikes and sharp-waves, predominantly in bilateral occipital regions. Genetic testing revealed heterozygous compound variants for a novel indel variant (c.116 - 117 delAG) and the repeat expansion of CSTB gene. The refractory BTCS and myoclonic jerks showed remarkable response to low-dose (2 mg/day) perampanel treatment. After 24 months of follow-up, the patient remained seizure-free, but her myoclonic jerks recurred, which could be reduced by increasing the dosage of perampanel.
Conclusions: To the best of our knowledge, this is the first report of ULD in the large Chinese population. By comparison with homozygous promoter expansions, we found an earlier age of first symptom onset and more refractory BTCS of ULD patients with compound or homozygous point/indel variants.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


