{"title":"Network insights into childhood obesity: unveiling methylated-differentially expressed genes and pathways through integrative bioinformatics analysis.","authors":"Felipe Mateus Pellenz, Guilherme Coutinho Kullmann Duarte, Giovanna Câmara Giudicelli, Thayne Woycinck Kowalski, Taís Silveira Assmann, Daisy Crispim","doi":"10.1530/EC-25-0049","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Childhood obesity, a global epidemic with profound impacts on physical and psychological health, remains a complex challenge with elusive underlying mechanisms. This study aimed to unravel the epigenetic landscape of this disease by identifying methylated-differentially expressed genes (MeDEGs) in childhood obesity through integrated bioinformatics approaches.</p><p><strong>Methods: </strong>Expression profiling (GSE9624) and methylation profiling (GSE25301, GSE27860, and GSE57484) datasets containing data on children with obesity (cases) and eutrophic children (control group) were obtained from the Gene Expression Omnibus (GEO) repository. Differentially expressed genes (DEGs) and differentially methylated genes (DMGs) between the groups were identified using GEO2R. MeDEGs were identified by superimposing the lists of DEGs and DMGs. The protein-protein interaction (PPI) network was constructed using the STRING database and analyzed using Cytoscape. Topological and modular PPI network analyses were carried out using the CytoHubba and MCODE plugins, respectively. Functional enrichment analyses were performed based on Gene Ontology terms and KEGG pathways.</p><p><strong>Results: </strong>A total of 70 MeDEGs were identified, including 45 hypomethylated high-expression and 25 hypermethylated low-expression genes. The PPI network highlighted three hub-bottleneck genes (CCL5, STAT1, and GATA3) and two functional modules. Overall, the 70 MeDEGs were associated with KEGG pathways related to cellular differentiation, inflammation, chemokine signaling, lipid and glucose metabolism, insulin resistance, and apoptosis.</p><p><strong>Conclusion: </strong>This study, employing integrative bioinformatics approaches, provides insights into the methylation-mediated mechanisms contributing to childhood obesity, advancing our understanding of this multifaceted chronic disease.</p>","PeriodicalId":11634,"journal":{"name":"Endocrine Connections","volume":" ","pages":""},"PeriodicalIF":2.8000,"publicationDate":"2025-06-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12147440/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Endocrine Connections","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1530/EC-25-0049","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/6/1 0:00:00","PubModel":"Print","JCR":"Q3","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Childhood obesity, a global epidemic with profound impacts on physical and psychological health, remains a complex challenge with elusive underlying mechanisms. This study aimed to unravel the epigenetic landscape of this disease by identifying methylated-differentially expressed genes (MeDEGs) in childhood obesity through integrated bioinformatics approaches.
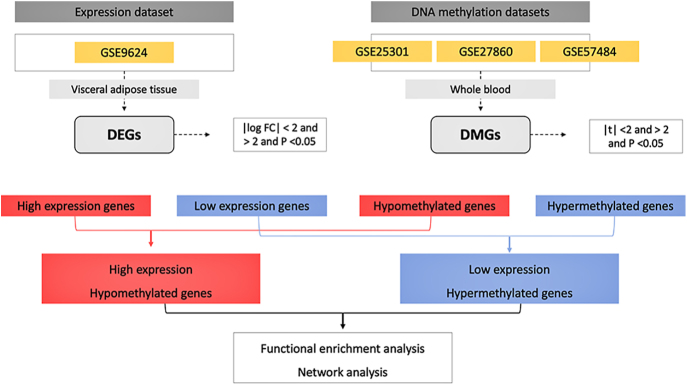
Methods: Expression profiling (GSE9624) and methylation profiling (GSE25301, GSE27860, and GSE57484) datasets containing data on children with obesity (cases) and eutrophic children (control group) were obtained from the Gene Expression Omnibus (GEO) repository. Differentially expressed genes (DEGs) and differentially methylated genes (DMGs) between the groups were identified using GEO2R. MeDEGs were identified by superimposing the lists of DEGs and DMGs. The protein-protein interaction (PPI) network was constructed using the STRING database and analyzed using Cytoscape. Topological and modular PPI network analyses were carried out using the CytoHubba and MCODE plugins, respectively. Functional enrichment analyses were performed based on Gene Ontology terms and KEGG pathways.
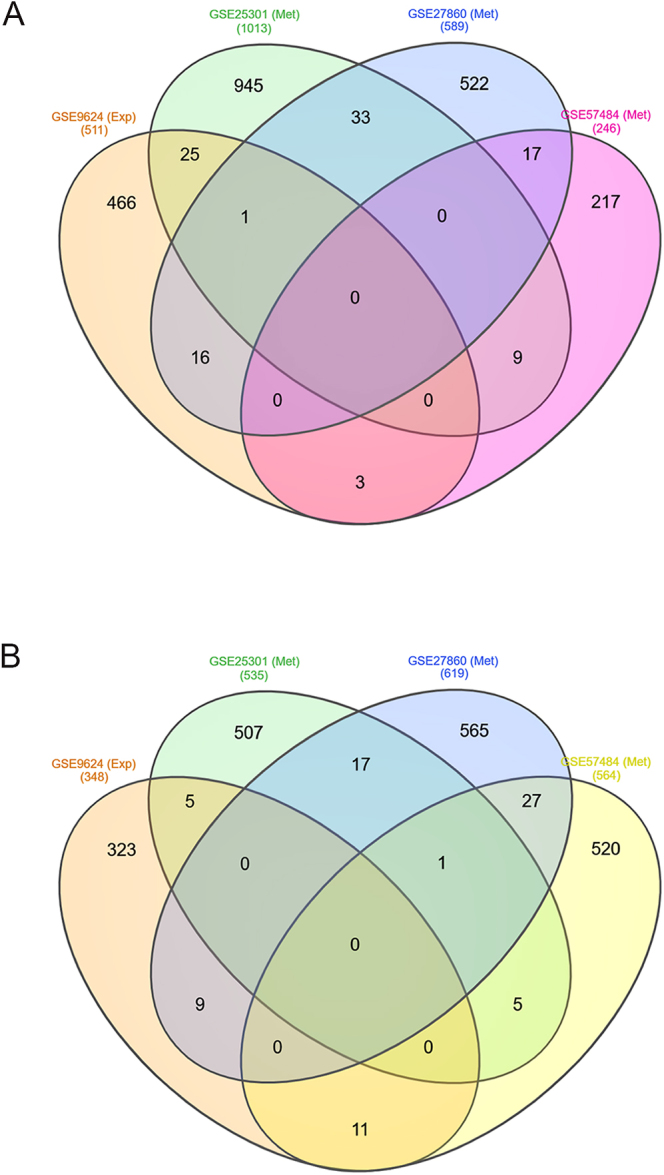
Results: A total of 70 MeDEGs were identified, including 45 hypomethylated high-expression and 25 hypermethylated low-expression genes. The PPI network highlighted three hub-bottleneck genes (CCL5, STAT1, and GATA3) and two functional modules. Overall, the 70 MeDEGs were associated with KEGG pathways related to cellular differentiation, inflammation, chemokine signaling, lipid and glucose metabolism, insulin resistance, and apoptosis.
Conclusion: This study, employing integrative bioinformatics approaches, provides insights into the methylation-mediated mechanisms contributing to childhood obesity, advancing our understanding of this multifaceted chronic disease.
背景:儿童肥胖是一种全球性的流行病,对身心健康有着深远的影响,是一个复杂的挑战,其潜在机制难以捉摸。本研究旨在通过综合生物信息学方法,通过鉴定儿童肥胖中的甲基化差异表达基因(MeDEGs),揭示这种疾病的表观遗传格局。方法:从Gene Expression Omnibus (GEO)知识库中获取包含肥胖儿童(病例)和富营养化儿童(对照组)数据的表达谱(GSE9624)和甲基化谱(GSE25301、GSE27860和GSE57484)数据集。采用GEO2R技术鉴定各组间差异表达基因(DEGs)和差异甲基化基因(DMGs)。通过将deg和dmg的列表叠加来识别megs。利用STRING数据库构建蛋白-蛋白相互作用(PPI)网络,并利用Cytoscape进行分析。分别使用CytoHubba和MCODE插件进行拓扑和模块化PPI网络分析。基于基因本体术语和KEGG通路进行功能富集分析。结果:共鉴定出70个medeg基因,其中低甲基化高表达基因45个,高甲基化低表达基因25个。PPI网络突出了三个枢纽瓶颈基因(CCL5、STAT1和GATA3)和两个功能模块。总体而言,70个medeg与细胞分化、炎症、趋化因子信号、脂质和葡萄糖代谢、胰岛素抵抗和细胞凋亡相关的KEGG通路相关。结论:本研究采用综合生物信息学方法,为甲基化介导的儿童肥胖机制提供了见解,促进了我们对这一多方面慢性疾病的理解。
期刊介绍:
Endocrine Connections publishes original quality research and reviews in all areas of endocrinology, including papers that deal with non-classical tissues as source or targets of hormones and endocrine papers that have relevance to endocrine-related and intersecting disciplines and the wider biomedical community.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


