Xianjun Han, Junxiang Cai, Can Bai* and Zijian Wu,
{"title":"Triview Molecular Representation Learning Combined with Multitask Optimization for Enhanced Molecular Property Prediction","authors":"Xianjun Han, Junxiang Cai, Can Bai* and Zijian Wu, ","doi":"10.1021/acs.jcim.5c0043610.1021/acs.jcim.5c00436","DOIUrl":null,"url":null,"abstract":"<p >In molecular property prediction tasks, most methods rely on single-view representations, such as simplified molecular input line entry system (SMILES) strings. Some scholars have attempted to combine two graphical views for joint representation purposes, such as SMILES and molecular graphs, but few have utilized three or more graphical views for molecular representation. Additionally, these methods typically extract features through pretraining models and then fine-tune them for specific tasks. This type of approach is not suitable for tasks with limited data and fails to fully leverage the correlations between tasks. To improve molecular representations, we propose a method that integrates traditional molecular representation learning by combining molecular sequences, molecular graphs, and molecular images. We design three different encoders to extract three graphical views of the same features from a molecule and use contrastive learning to align these views. Moreover, we adopt a multitask optimization strategy that effectively utilizes the shared information and correlations between tasks, thereby improving the generalizability and predictive performance of the model. Finally, we use low-rank adaptation (LoRA) fine-tuning for specific tasks to further improve the output prediction results. The experimental results show that this method enhances the accuracy and robustness of molecular property prediction across multiple benchmark data sets.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"65 10","pages":"5163–5175 5163–5175"},"PeriodicalIF":5.3000,"publicationDate":"2025-05-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jcim.5c00436","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
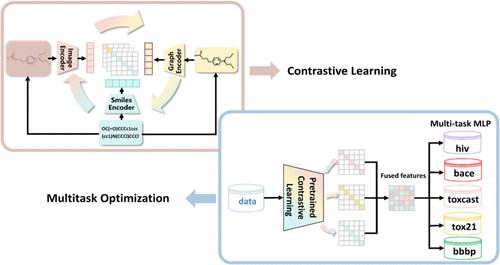
In molecular property prediction tasks, most methods rely on single-view representations, such as simplified molecular input line entry system (SMILES) strings. Some scholars have attempted to combine two graphical views for joint representation purposes, such as SMILES and molecular graphs, but few have utilized three or more graphical views for molecular representation. Additionally, these methods typically extract features through pretraining models and then fine-tune them for specific tasks. This type of approach is not suitable for tasks with limited data and fails to fully leverage the correlations between tasks. To improve molecular representations, we propose a method that integrates traditional molecular representation learning by combining molecular sequences, molecular graphs, and molecular images. We design three different encoders to extract three graphical views of the same features from a molecule and use contrastive learning to align these views. Moreover, we adopt a multitask optimization strategy that effectively utilizes the shared information and correlations between tasks, thereby improving the generalizability and predictive performance of the model. Finally, we use low-rank adaptation (LoRA) fine-tuning for specific tasks to further improve the output prediction results. The experimental results show that this method enhances the accuracy and robustness of molecular property prediction across multiple benchmark data sets.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


