Ana María Velásquez-Escobar, Andrew E Hillhouse, Terry Magnuson, David W Threadgill
{"title":"Snrnp25 is a candidate for the peri-implantation lethal phenotype of the Hba deletions.","authors":"Ana María Velásquez-Escobar, Andrew E Hillhouse, Terry Magnuson, David W Threadgill","doi":"10.1007/s00335-025-10133-z","DOIUrl":null,"url":null,"abstract":"<p><p>Mutations in adult hemoglobin alpha genes in humans lead to blood disorders commonly known as α-thalassemia. In search of a mouse model for this disease, mutagenesis screens have identified several deletions that resemble these phenotypes. The Hba<sup>b2(th)</sup> deletion, in particular, replicates the characteristics of alpha-thalassemia minor in heterozygous mice but presents a homozygous embryonic lethal phenotype. Previous analyses of Hba<sup>b2(th)</sup> mice suggested that the deletion affects both Hba genes (Hba-a1 and Hba-a2) and considered epidermal growth factor receptor (Egfr) or rhomboid 5 homolog 1 (Rhbdf1) to be responsible for the embryonic lethality. Molecular analysis of Hba<sup>b2(th)</sup> revealed a deletion spanning a 1 cM region of mouse chromosome 11. Importantly, the Hba<sup>b2(th)</sup> deletion does not extend to Egfr, indicating that the observed lethality of homozygous embryos is not due to the loss of Egfr. Sequence analysis of the Hba<sup>b2(th)</sup> deletion showed that the Hba-a2 gene is not deleted, but the lack of expression is likely due to the disruption of upstream regulatory regions. Furthermore, we identify Snrnp25, which codes for the small nuclear ribonucleoprotein 25 (U11/U12), as the candidate gene most likely responsible for the peri-implantation lethality of Hba<sup>b2(th)</sup> homozygous mice. These findings enhance the understanding of the genetic mechanisms underlying α-thalassemia and provide insights into novel genes essential for early mammalian development.</p>","PeriodicalId":18259,"journal":{"name":"Mammalian Genome","volume":" ","pages":"727-734"},"PeriodicalIF":2.7000,"publicationDate":"2025-09-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12408735/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Mammalian Genome","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00335-025-10133-z","RegionNum":4,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/21 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
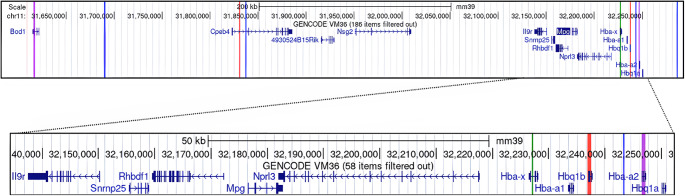
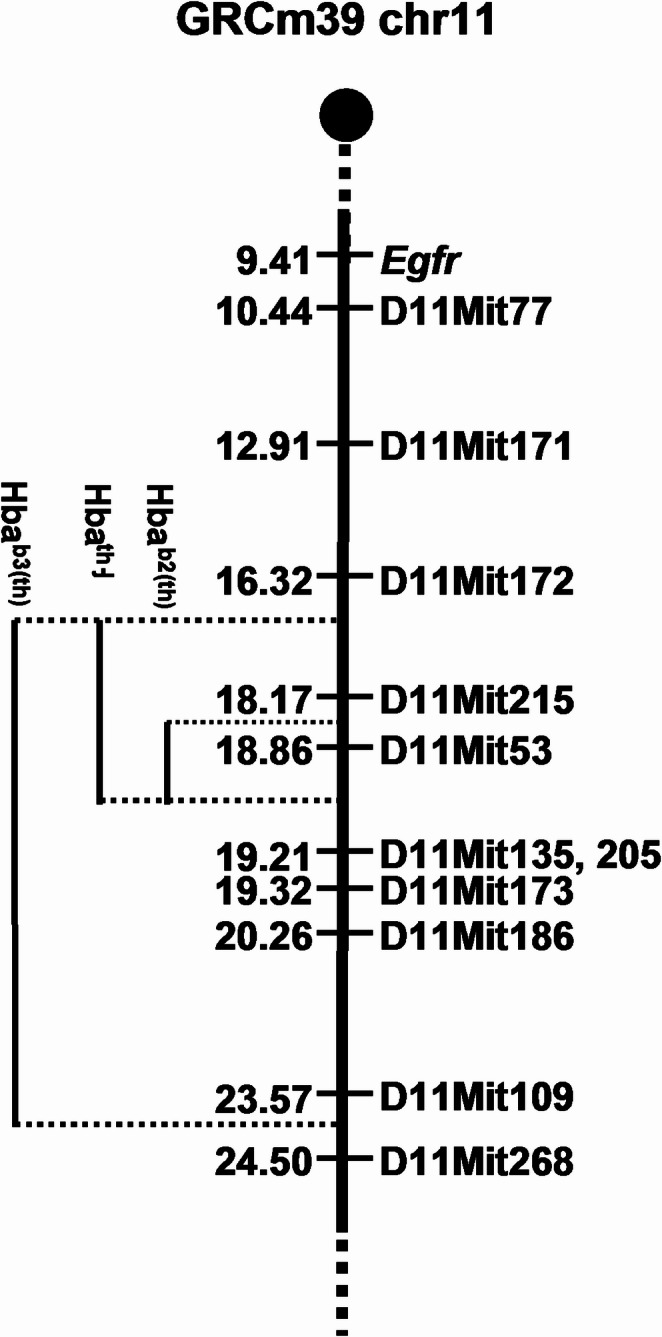
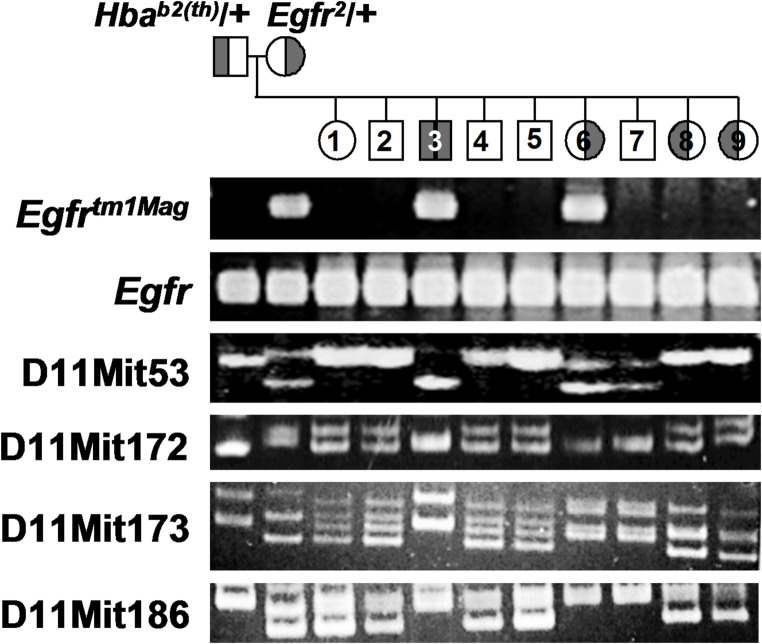
Mutations in adult hemoglobin alpha genes in humans lead to blood disorders commonly known as α-thalassemia. In search of a mouse model for this disease, mutagenesis screens have identified several deletions that resemble these phenotypes. The Hbab2(th) deletion, in particular, replicates the characteristics of alpha-thalassemia minor in heterozygous mice but presents a homozygous embryonic lethal phenotype. Previous analyses of Hbab2(th) mice suggested that the deletion affects both Hba genes (Hba-a1 and Hba-a2) and considered epidermal growth factor receptor (Egfr) or rhomboid 5 homolog 1 (Rhbdf1) to be responsible for the embryonic lethality. Molecular analysis of Hbab2(th) revealed a deletion spanning a 1 cM region of mouse chromosome 11. Importantly, the Hbab2(th) deletion does not extend to Egfr, indicating that the observed lethality of homozygous embryos is not due to the loss of Egfr. Sequence analysis of the Hbab2(th) deletion showed that the Hba-a2 gene is not deleted, but the lack of expression is likely due to the disruption of upstream regulatory regions. Furthermore, we identify Snrnp25, which codes for the small nuclear ribonucleoprotein 25 (U11/U12), as the candidate gene most likely responsible for the peri-implantation lethality of Hbab2(th) homozygous mice. These findings enhance the understanding of the genetic mechanisms underlying α-thalassemia and provide insights into novel genes essential for early mammalian development.
期刊介绍:
Mammalian Genome focuses on the experimental, theoretical and technical aspects of genetics, genomics, epigenetics and systems biology in mouse, human and other mammalian species, with an emphasis on the relationship between genotype and phenotype, elucidation of biological and disease pathways as well as experimental aspects of interventions, therapeutics, and precision medicine. The journal aims to publish high quality original papers that present novel findings in all areas of mammalian genetic research as well as review articles on areas of topical interest. The journal will also feature commentaries and editorials to inform readers of breakthrough discoveries as well as issues of research standards, policies and ethics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


