Syndromic forms of inherited retinal dystrophies: a comprehensive molecular diagnosis of consanguineous Pakistani families using capture panel sequencing.
IF 1.4 3区 医学Q4 BIOCHEMISTRY & MOLECULAR BIOLOGY
Molecular VisionPub Date : 2025-03-26eCollection Date: 2025-01-01
{"title":"Syndromic forms of inherited retinal dystrophies: a comprehensive molecular diagnosis of consanguineous Pakistani families using capture panel sequencing.","authors":"Aleesha Asghar, Sumbal Wazir, Shehzeen Fatima, Hussan Bilal, Muhammad Shoaib, Saqib Ur Rehman, Sumaira Altaf, Yumei Li, Kiran Afshan, Rui Chen, Sabika Firasat","doi":"","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Inherited retinal dystrophies (IRDs) represent a clinically and genetically heterogeneous group of genetic disorders that involve photoreceptors and/or retinal pigment epithelium degeneration. IRDs may occur as an isolated condition or may represent an ocular manifestation of a multisystemic disorder referred as syndromic IRD. To increase the understanding of the molecular determinants of syndromic IRD-related genes in the Pakistani population, we revealed the genetic profile of 13 consanguineous Pakistani families using capture panel sequencing.</p><p><strong>Methods: </strong>We performed comprehensive molecular testing on 72 IRD segregating Pakistani families using targeted capture panel sequencing of 344 known genes. The pathogenicity of candidate variants was assessed using American College of Medical Genetics and Genomics guidelines, followed by Sanger sequencing for segregation analysis.</p><p><strong>Results: </strong>Causative variants in previously reported syndromic IRDs genes were detected in 13/72 (18%) IRD families, including 5/72 (6.94%), 4/72 (5.55%), 2/72 (2.8%), 1/72(1.38%) and 1/72 (1.38%) in Usher syndrome, Bardet-Biedl syndrome, Batten disease, retinitis pigmentosa with situs inversus and Stickler syndrome segregated families, respectively. Disease-causing variants included nine previously reported and six novel homozygous variants, i.e., c.1143G>C in <i>USH2A</i>, c.470G>A in <i>MYO7A</i>, c.877-2A>G in <i>PCDH15</i>, c.347C>T in <i>ARL6</i>, c.581C>T in <i>CLN5</i> and c.100+1G>T in <i>ARL2BP</i> gene segregation with disease phenotype in eight families. Two heterozygous variants of the <i>USH2A</i> gene, i.e., c.12093C>A and c.9815C>T, were segregated in a compound heterozygous form in family RP243. Furthermore, RP151 showed segregation of a heterozygous variant c.247G>A in a Stickler syndrome gene, i.e., <i>COL2A1</i>, in an autosomal dominant manner.</p><p><strong>Conclusions: </strong>This study reaffirms the clinical and genetic heterogeneity of syndromic IRD-associated genes and confirms the usefulness of molecular methods in advancing our understanding of these conditions in consanguineous populations. The most commonly mutated Bardet-Biedl syndrome gene was <i>ARL6</i> (75%) and the most commonly mutated Usher syndrome genes were <i>USH2A</i> (40%) and <i>MYO7A</i> (40%). Our data could serve as a reference for future studies and the development of treatment modalities for affected families of Pakistani origin.</p>","PeriodicalId":18866,"journal":{"name":"Molecular Vision","volume":"31 ","pages":"69-83"},"PeriodicalIF":1.4000,"publicationDate":"2025-03-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12085215/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Vision","FirstCategoryId":"3","ListUrlMain":"","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Inherited retinal dystrophies (IRDs) represent a clinically and genetically heterogeneous group of genetic disorders that involve photoreceptors and/or retinal pigment epithelium degeneration. IRDs may occur as an isolated condition or may represent an ocular manifestation of a multisystemic disorder referred as syndromic IRD. To increase the understanding of the molecular determinants of syndromic IRD-related genes in the Pakistani population, we revealed the genetic profile of 13 consanguineous Pakistani families using capture panel sequencing.
Methods: We performed comprehensive molecular testing on 72 IRD segregating Pakistani families using targeted capture panel sequencing of 344 known genes. The pathogenicity of candidate variants was assessed using American College of Medical Genetics and Genomics guidelines, followed by Sanger sequencing for segregation analysis.
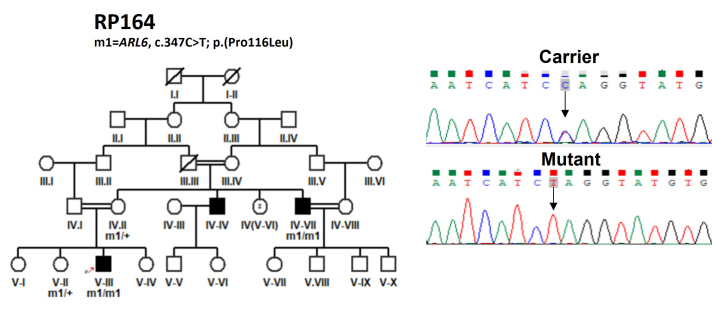
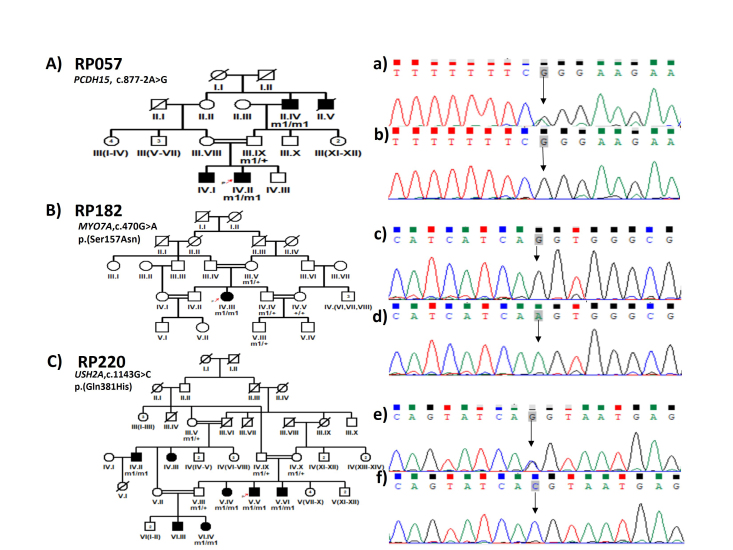
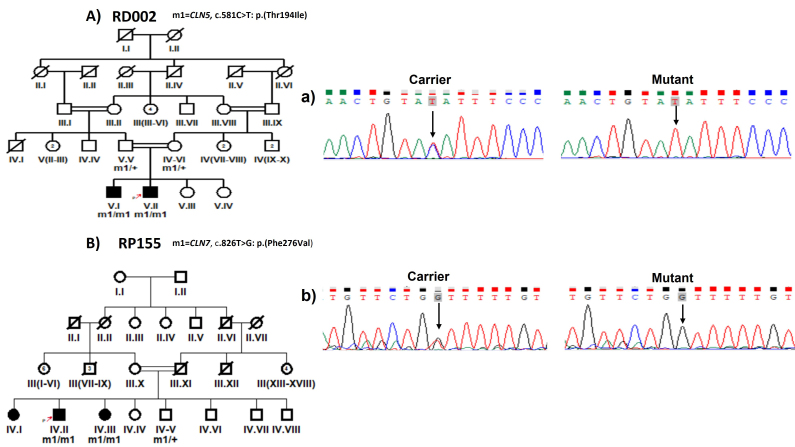
Results: Causative variants in previously reported syndromic IRDs genes were detected in 13/72 (18%) IRD families, including 5/72 (6.94%), 4/72 (5.55%), 2/72 (2.8%), 1/72(1.38%) and 1/72 (1.38%) in Usher syndrome, Bardet-Biedl syndrome, Batten disease, retinitis pigmentosa with situs inversus and Stickler syndrome segregated families, respectively. Disease-causing variants included nine previously reported and six novel homozygous variants, i.e., c.1143G>C in USH2A, c.470G>A in MYO7A, c.877-2A>G in PCDH15, c.347C>T in ARL6, c.581C>T in CLN5 and c.100+1G>T in ARL2BP gene segregation with disease phenotype in eight families. Two heterozygous variants of the USH2A gene, i.e., c.12093C>A and c.9815C>T, were segregated in a compound heterozygous form in family RP243. Furthermore, RP151 showed segregation of a heterozygous variant c.247G>A in a Stickler syndrome gene, i.e., COL2A1, in an autosomal dominant manner.
Conclusions: This study reaffirms the clinical and genetic heterogeneity of syndromic IRD-associated genes and confirms the usefulness of molecular methods in advancing our understanding of these conditions in consanguineous populations. The most commonly mutated Bardet-Biedl syndrome gene was ARL6 (75%) and the most commonly mutated Usher syndrome genes were USH2A (40%) and MYO7A (40%). Our data could serve as a reference for future studies and the development of treatment modalities for affected families of Pakistani origin.
期刊介绍:
Molecular Vision is a peer-reviewed journal dedicated to the dissemination of research results in molecular biology, cell biology, and the genetics of the visual system (ocular and cortical).
Molecular Vision publishes articles presenting original research that has not previously been published and comprehensive articles reviewing the current status of a particular field or topic. Submissions to Molecular Vision are subjected to rigorous peer review. Molecular Vision does NOT publish preprints.
For authors, Molecular Vision provides a rapid means of communicating important results. Access to Molecular Vision is free and unrestricted, allowing the widest possible audience for your article. Digital publishing allows you to use color images freely (and without fees). Additionally, you may publish animations, sounds, or other supplementary information that clarifies or supports your article. Each of the authors of an article may also list an electronic mail address (which will be updated upon request) to give interested readers easy access to authors.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


