Calculation of Single Molecule Conductance from Molecular Dynamics Simulations: Implementation in the SIESTA Code
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
The high computational cost of computing the conductance of single molecule junctions with standard methods based on Density-Functional Theory and Non-Equilibrium Green’s Functions (DFT-NEGF) limits its application to only a few junction geometries. Thus, calculations probing the variability of conductance using geometries from Molecular Dynamics simulations must often resort to approximate descriptions of the electronic structure. We describe the implementation in the SIESTA code of a method that allows for the fast computation of junction conductance from Molecular Dynamics geometries without a simplified description of the electronic structure. Its efficiency comes from considering small Au-molecule-Au clusters, which properly capture variations in conductance arising from changes in molecular conformation. After scaling with a few DFT-NEGF calculations, the conductance of tens of thousands of junction geometries can be accurately computed at the DFT level more than 2 orders of magnitude faster than with DFT-NEGF. Coupled with MD simulations at room temperature, it is used here to quantify the role of benzene ring rotations in conductance using the ensemble of geometries from the junction dynamics in paradigmatic single molecule junctions.
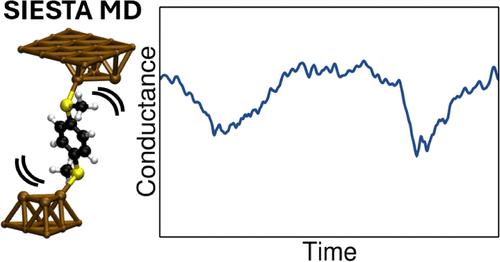
分子动力学模拟中单分子电导的计算:在SIESTA代码中实现
基于密度泛函理论和非平衡格林函数(DFT-NEGF)的标准方法计算单分子结电导的计算成本高,限制了其仅适用于少数结几何。因此,利用分子动力学模拟的几何图形探测电导可变性的计算必须经常求助于电子结构的近似描述。我们在SIESTA代码中描述了一种方法的实现,该方法允许从分子动力学几何中快速计算结电导,而无需简化电子结构的描述。它的效率来自于考虑小的au分子- au团簇,它适当地捕获了由分子构象变化引起的电导变化。经过几次DFT- negf计算后,数万个结几何形状的电导可以在DFT水平上精确计算,比使用DFT- negf快2个数量级以上。结合室温下的分子动力学模拟,本文使用聚合单分子结动力学的几何集合来量化苯环旋转在电导中的作用。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


