{"title":"Progranulin deficiency in the brain: the interplay between neuronal and non-neuronal cells.","authors":"Katarzyna Gaweda-Walerych, Vanessa Aragona, Simona Lodato, Emilia J Sitek, Ewa Narożańska, Emanuele Buratti","doi":"10.1186/s40035-025-00475-8","DOIUrl":null,"url":null,"abstract":"<p><p>Heterozygous mutations in GRN gene lead to insufficient levels of the progranulin (PGRN) protein, resulting in frontotemporal dementia (FTD) with TAR DNA-binding protein 43 (TDP-43) inclusions, classified pathologically as frontotemporal lobar degeneration (FTLD-TDP). Homozygous GRN mutations are exceedingly rare and cause neuronal ceroid lipofuscinosis 11, a lysosomal storage disease with onset in young adulthood, or an FTD syndrome with late-onset manifestations. In this review, we highlight the broad spectrum of clinical phenotypes associated with PGRN deficiency, including primary progressive aphasia and behavioral variant of frontotemporal dementia. We explore these phenotypes alongside relevant rodent and in vitro human models, ranging from the induced pluripotent stem cell-derived neural progenitors, neurons, microglia, and astrocytes to genetically engineered heterotypic organoids containing both neurons and astrocytes. We summarize advantages and limitations of these models in recapitulating the main FTLD-GRN hallmarks, highlighting the role of non-cell-autonomous mechanisms in the formation of TDP-43 pathology, neuroinflammation, and neurodegeneration. Data obtained from patients' brain tissues and biofluids, in parallel with single-cell transcriptomics, demonstrate the complexity of interactions among the highly heterogeneous cellular clusters present in the brain, including neurons, astrocytes, microglia, oligodendroglia, endothelial cells, and pericytes. Emerging evidence has revealed that PGRN deficiency is associated with cell cluster-specific, often conserved, genetic and molecular phenotypes in the central nervous system. In this review, we focus on how these distinct cellular populations and their dysfunctional crosstalk contribute to neurodegeneration and neuroinflammation in FTD-GRN. Specifically, we characterize the phenotypes of lipid droplet-accumulating microglia and alterations of myelin lipid content resulting from lysosomal dysfunction caused by PGRN deficiency. Additionally, we consider how the deregulation of glia-neuron communication affects the exchange of organelles such as mitochondria, and the removal of excess toxic products such as protein aggregates, in PGRN-related neurodegeneration.</p>","PeriodicalId":23269,"journal":{"name":"Translational Neurodegeneration","volume":"14 1","pages":"18"},"PeriodicalIF":15.2000,"publicationDate":"2025-04-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12001433/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational Neurodegeneration","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s40035-025-00475-8","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"NEUROSCIENCES","Score":null,"Total":0}
引用次数: 0
Abstract
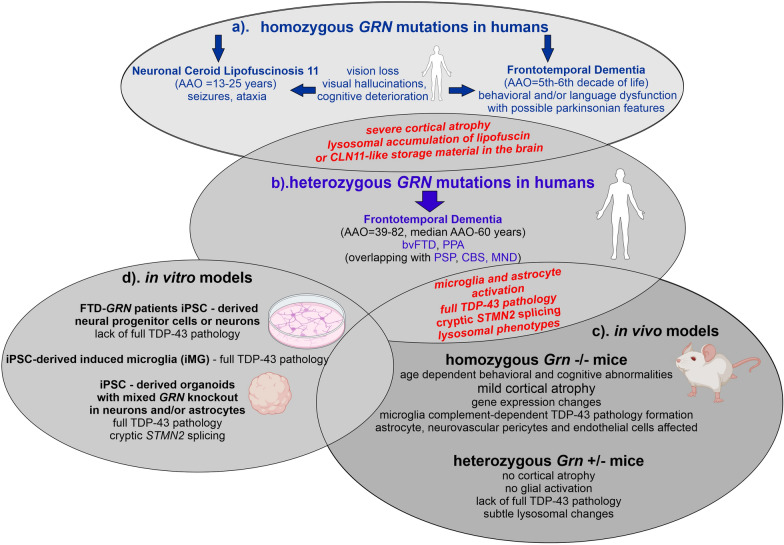
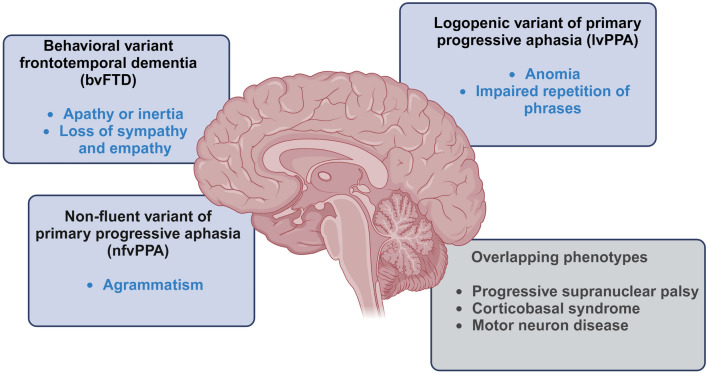
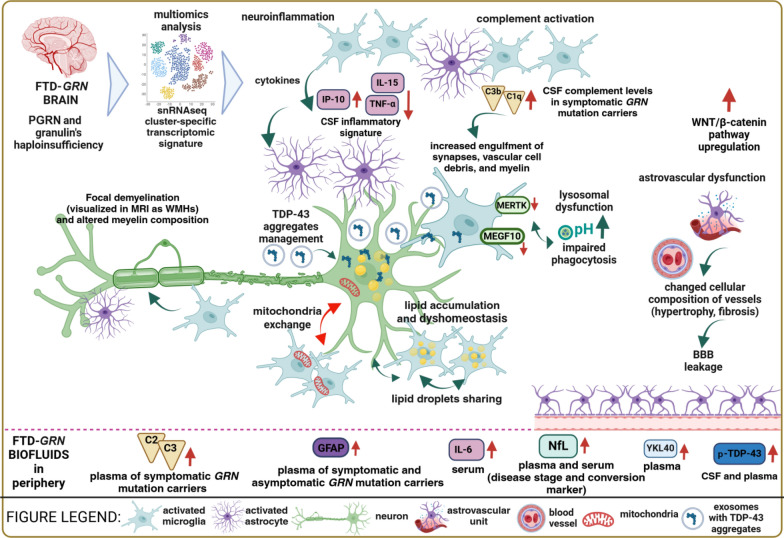
Heterozygous mutations in GRN gene lead to insufficient levels of the progranulin (PGRN) protein, resulting in frontotemporal dementia (FTD) with TAR DNA-binding protein 43 (TDP-43) inclusions, classified pathologically as frontotemporal lobar degeneration (FTLD-TDP). Homozygous GRN mutations are exceedingly rare and cause neuronal ceroid lipofuscinosis 11, a lysosomal storage disease with onset in young adulthood, or an FTD syndrome with late-onset manifestations. In this review, we highlight the broad spectrum of clinical phenotypes associated with PGRN deficiency, including primary progressive aphasia and behavioral variant of frontotemporal dementia. We explore these phenotypes alongside relevant rodent and in vitro human models, ranging from the induced pluripotent stem cell-derived neural progenitors, neurons, microglia, and astrocytes to genetically engineered heterotypic organoids containing both neurons and astrocytes. We summarize advantages and limitations of these models in recapitulating the main FTLD-GRN hallmarks, highlighting the role of non-cell-autonomous mechanisms in the formation of TDP-43 pathology, neuroinflammation, and neurodegeneration. Data obtained from patients' brain tissues and biofluids, in parallel with single-cell transcriptomics, demonstrate the complexity of interactions among the highly heterogeneous cellular clusters present in the brain, including neurons, astrocytes, microglia, oligodendroglia, endothelial cells, and pericytes. Emerging evidence has revealed that PGRN deficiency is associated with cell cluster-specific, often conserved, genetic and molecular phenotypes in the central nervous system. In this review, we focus on how these distinct cellular populations and their dysfunctional crosstalk contribute to neurodegeneration and neuroinflammation in FTD-GRN. Specifically, we characterize the phenotypes of lipid droplet-accumulating microglia and alterations of myelin lipid content resulting from lysosomal dysfunction caused by PGRN deficiency. Additionally, we consider how the deregulation of glia-neuron communication affects the exchange of organelles such as mitochondria, and the removal of excess toxic products such as protein aggregates, in PGRN-related neurodegeneration.
期刊介绍:
Translational Neurodegeneration, an open-access, peer-reviewed journal, addresses all aspects of neurodegenerative diseases. It serves as a prominent platform for research, therapeutics, and education, fostering discussions and insights across basic, translational, and clinical research domains. Covering Parkinson's disease, Alzheimer's disease, and other neurodegenerative conditions, it welcomes contributions on epidemiology, pathogenesis, diagnosis, prevention, drug development, rehabilitation, and drug delivery. Scientists, clinicians, and physician-scientists are encouraged to share their work in this specialized journal tailored to their fields.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


