Hien Bao Dieu Thai, WonWoo Jung, Sol Choi, Woo Joong Kim, JangSup Moon, ByungChan Lim
{"title":"Establishing an induced pluripotent stem cell bank using urine cells from pediatric patients with neurogenetic diseases.","authors":"Hien Bao Dieu Thai, WonWoo Jung, Sol Choi, Woo Joong Kim, JangSup Moon, ByungChan Lim","doi":"10.3345/cep.2024.01830","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Inadequate knowledge of the fundamental mechanisms underlying pediatric neurological disorders impedes their effective treatment. Induced pluripotent stem cells (iPSCs) are essential for exploring the course of neurological diseases because they enable disease modeling at the cellular level.</p><p><strong>Purpose: </strong>This study aimed to generate an iPSC bank using urine cells (UCs) for clinical applications, particularly the study of pediatric neurogenetic diseases. Urine sample collections can benefit a large donor population because they use a noninvasive, painless, and simple technique that provides plentiful cells for iPSC generation.</p><p><strong>Methods: </strong>UCs were isolated from the urine of donors with specific diseases (n=12; 7 males, 5 females). The UCs were reprogrammed into iPSCs using episomal plasmid vectors and key transcription factors (OCT3/4, SOX2, KLF4, L-MYC, and LIN28). Quantitative polymerase chain reaction and immunocytochemical analyses confirmed the expression of pluripotent genes (OCT3/4, SOX2, NANOG, and LIN28) and proteins (OCT4, NANOG, SSEA-4, and TRA-1-60). Trilineage differentiation was investigated by immunostaining embryonic body-derived iPSCs for β-tubulin III, smooth muscle actin, and alpha-fetoprotein. The genomic stability of the iPSCs was assessed using chromosomal microarray (CMA).</p><p><strong>Results: </strong>UCs were successfully isolated from patients with various early-onset neurogenetic diseases and reprogrammed into iPSCs. The iPSCs were confirmed as pluripotent and capable of trilineage differentiation as evidenced by the enhanced expression of relevant genes and proteins. The genomic profiles of the iPSCs were assessed using CMA, which revealed that 4 of the 12 lines exhibited pathogenic chromosomal deletions or duplications. Interestingly, repeated CMA tests using earlier-passage cells resulted in normal findings in one of the 4 iPSC lines. These findings highlight the need for genetic screening throughout the culture period.</p><p><strong>Conclusion: </strong>Here we used UCs to successfully develop an early-onset neurogenetic disease iPSC bank that offers an efficient protocol for expanding patient accessibility in pediatric neurogenetic research.</p>","PeriodicalId":36018,"journal":{"name":"Clinical and Experimental Pediatrics","volume":" ","pages":"569-577"},"PeriodicalIF":3.6000,"publicationDate":"2025-08-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12326042/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical and Experimental Pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3345/cep.2024.01830","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/4/1 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PEDIATRICS","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Inadequate knowledge of the fundamental mechanisms underlying pediatric neurological disorders impedes their effective treatment. Induced pluripotent stem cells (iPSCs) are essential for exploring the course of neurological diseases because they enable disease modeling at the cellular level.
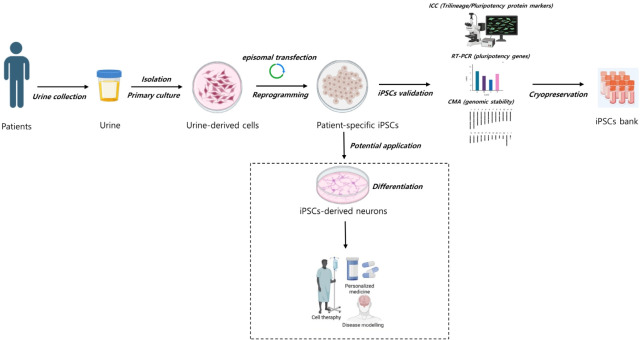
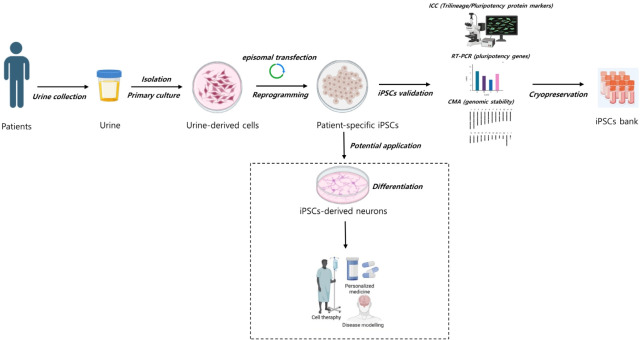
Purpose: This study aimed to generate an iPSC bank using urine cells (UCs) for clinical applications, particularly the study of pediatric neurogenetic diseases. Urine sample collections can benefit a large donor population because they use a noninvasive, painless, and simple technique that provides plentiful cells for iPSC generation.
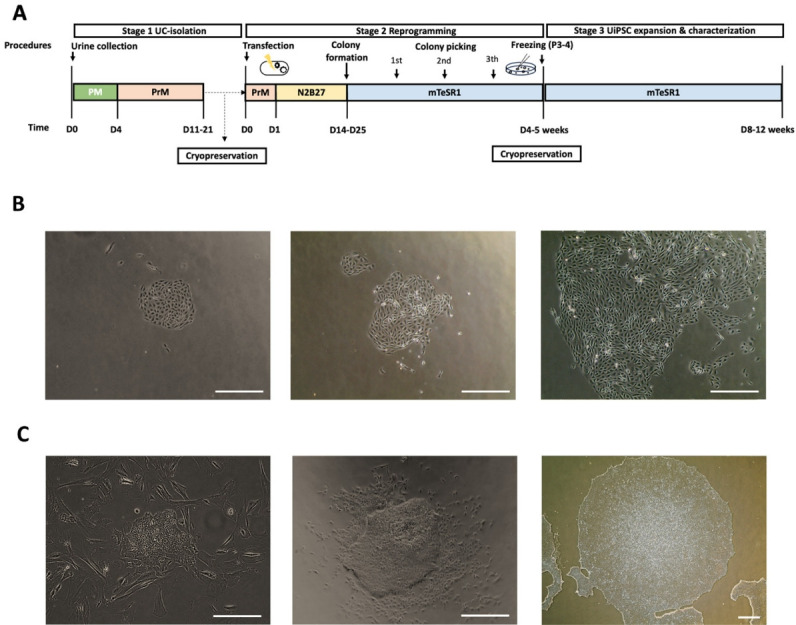
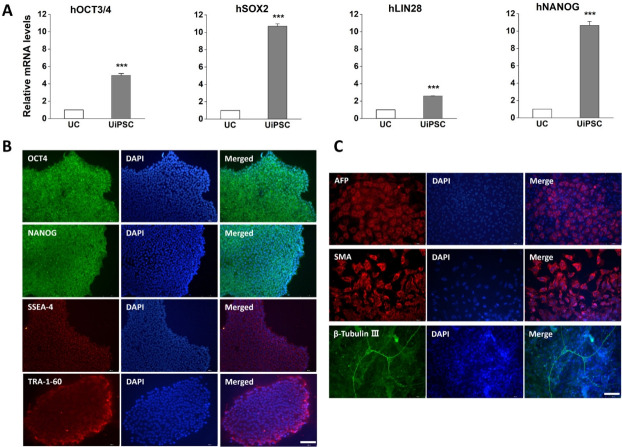
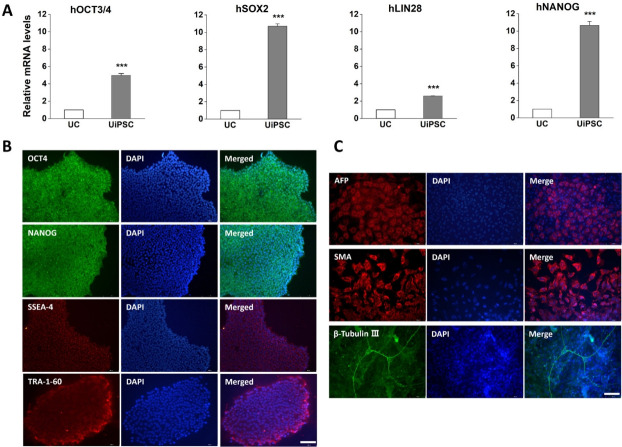
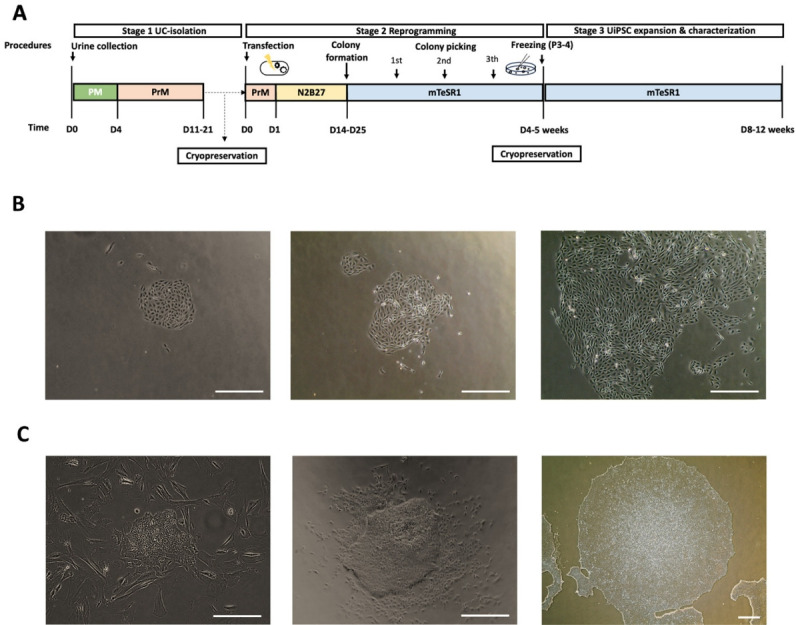
Methods: UCs were isolated from the urine of donors with specific diseases (n=12; 7 males, 5 females). The UCs were reprogrammed into iPSCs using episomal plasmid vectors and key transcription factors (OCT3/4, SOX2, KLF4, L-MYC, and LIN28). Quantitative polymerase chain reaction and immunocytochemical analyses confirmed the expression of pluripotent genes (OCT3/4, SOX2, NANOG, and LIN28) and proteins (OCT4, NANOG, SSEA-4, and TRA-1-60). Trilineage differentiation was investigated by immunostaining embryonic body-derived iPSCs for β-tubulin III, smooth muscle actin, and alpha-fetoprotein. The genomic stability of the iPSCs was assessed using chromosomal microarray (CMA).
Results: UCs were successfully isolated from patients with various early-onset neurogenetic diseases and reprogrammed into iPSCs. The iPSCs were confirmed as pluripotent and capable of trilineage differentiation as evidenced by the enhanced expression of relevant genes and proteins. The genomic profiles of the iPSCs were assessed using CMA, which revealed that 4 of the 12 lines exhibited pathogenic chromosomal deletions or duplications. Interestingly, repeated CMA tests using earlier-passage cells resulted in normal findings in one of the 4 iPSC lines. These findings highlight the need for genetic screening throughout the culture period.
Conclusion: Here we used UCs to successfully develop an early-onset neurogenetic disease iPSC bank that offers an efficient protocol for expanding patient accessibility in pediatric neurogenetic research.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


