Djazia Haferssas, Marion Dubuissez, Jonatan Barrera-Chimal, Clémence Messmer, El Bachir Affar, Bruno Larrivée, Xue-Song Liu, Casimiro Gerarduzzi
{"title":"FLT4 activation promotes acute lymphoid leukemia survival through stabilization of MDM2/MDMX and inactivation of p53.","authors":"Djazia Haferssas, Marion Dubuissez, Jonatan Barrera-Chimal, Clémence Messmer, El Bachir Affar, Bruno Larrivée, Xue-Song Liu, Casimiro Gerarduzzi","doi":"10.1038/s41389-025-00552-7","DOIUrl":null,"url":null,"abstract":"<p><p>Aberrant Receptor Tyrosine Kinase (RTK) signaling allows cancer cells to modulate survival, proliferation, and death, leading to tumorigenesis and chemoresistance. In leukemia, the RTK FMS-Related Tyrosine Kinase 4 (FLT4) (also known as VEGFR3, Vascular Endothelial Growth Factor Receptor- 3) is deregulated and correlates with cancer progression. However, the underlying consequences of its deregulation remain to be determined. Moreover, chemotherapy treatment requires that cancer cells retain a wild-type p53 to respond to DNA damage by tumor-suppressing activities, i.e. apoptosis. p53 activity is predominantly limited by its two major negative regulators, MDM2 and MDMX, which inactivate p53 by promoting its degradation and/or cytoplasmic localization. In this study, we have shown that activation of FLT4 by either overexpression or binding of its ligand, VEGFC, increases MDM2/MDMX stability, inactivates p53, and leads to resistance to DNA-damaging therapies. Moreover, we found that MDMX Ser-314 phosphorylation, a consensus sequence of CDK4/6, increases MDMX stability, which subsequently affects MDM2 and p53 degradation and could be reversed by the CDK4/6 inhibitor Palbociclib. More importantly, leukemic cells treated with Palbociclib were more susceptible to DNA-damaging induction of apoptosis and had reduced cell proliferation. Leukemic cells overexpressing FLT4 displayed accelerated proliferation when injected into NOD-SCID mice as compared to wild-type cells. Altogether, our research proposes an innovative way to reactivate p53 in leukemia through the pharmacological inhibition of FLT4 signaling, which could serve as a potential treatment option. Schematic representation of FLT4-mediated MDM2/MDMX complex stabilization and suppression of p53 activity. VEGFC triggers FLT4 activation, leading to CDK4/6 activation, which phosphorylates MDMX on Ser-314. As a result, MDMX levels increase and bind to MDM2, stabilizing the MDM2/MDMX complex. This complex binds to p53, facilitating its suppression by reducing its transcriptional activity or enhancing its export to the cytoplasm for proteasomal degradation. Consequently, p53 inactivation promotes their survival, proliferation, and resistance to chemotherapy-induced apoptosis. The figure was created in BioRender.com.</p>","PeriodicalId":19489,"journal":{"name":"Oncogenesis","volume":"14 1","pages":"14"},"PeriodicalIF":6.4000,"publicationDate":"2025-05-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12048674/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Oncogenesis","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41389-025-00552-7","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
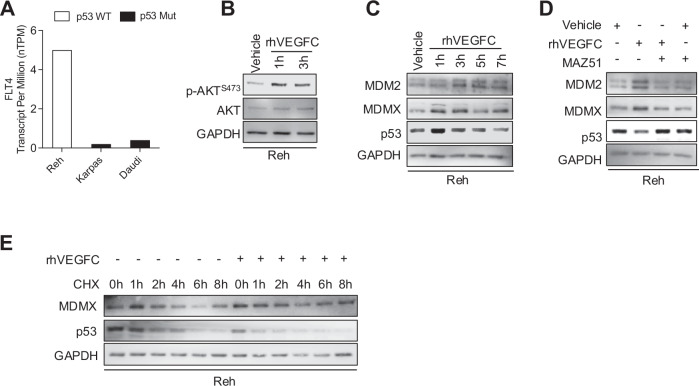
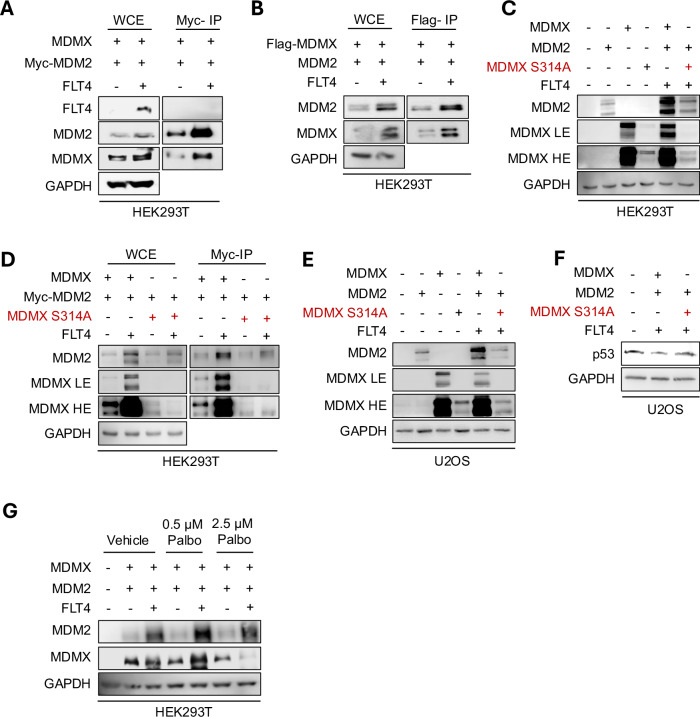
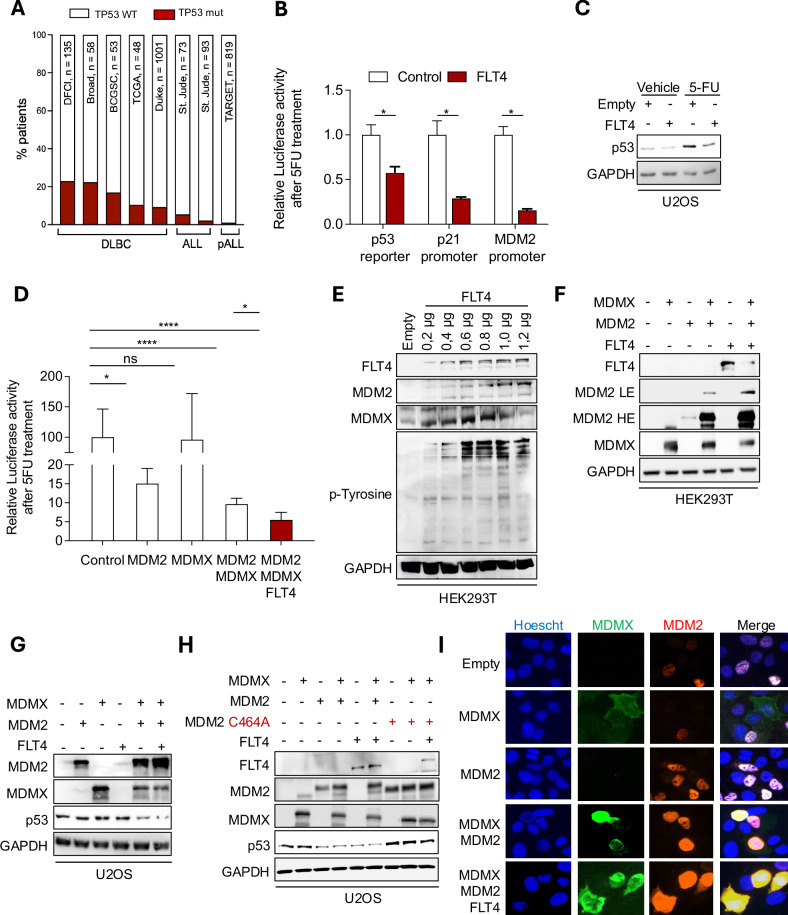
Aberrant Receptor Tyrosine Kinase (RTK) signaling allows cancer cells to modulate survival, proliferation, and death, leading to tumorigenesis and chemoresistance. In leukemia, the RTK FMS-Related Tyrosine Kinase 4 (FLT4) (also known as VEGFR3, Vascular Endothelial Growth Factor Receptor- 3) is deregulated and correlates with cancer progression. However, the underlying consequences of its deregulation remain to be determined. Moreover, chemotherapy treatment requires that cancer cells retain a wild-type p53 to respond to DNA damage by tumor-suppressing activities, i.e. apoptosis. p53 activity is predominantly limited by its two major negative regulators, MDM2 and MDMX, which inactivate p53 by promoting its degradation and/or cytoplasmic localization. In this study, we have shown that activation of FLT4 by either overexpression or binding of its ligand, VEGFC, increases MDM2/MDMX stability, inactivates p53, and leads to resistance to DNA-damaging therapies. Moreover, we found that MDMX Ser-314 phosphorylation, a consensus sequence of CDK4/6, increases MDMX stability, which subsequently affects MDM2 and p53 degradation and could be reversed by the CDK4/6 inhibitor Palbociclib. More importantly, leukemic cells treated with Palbociclib were more susceptible to DNA-damaging induction of apoptosis and had reduced cell proliferation. Leukemic cells overexpressing FLT4 displayed accelerated proliferation when injected into NOD-SCID mice as compared to wild-type cells. Altogether, our research proposes an innovative way to reactivate p53 in leukemia through the pharmacological inhibition of FLT4 signaling, which could serve as a potential treatment option. Schematic representation of FLT4-mediated MDM2/MDMX complex stabilization and suppression of p53 activity. VEGFC triggers FLT4 activation, leading to CDK4/6 activation, which phosphorylates MDMX on Ser-314. As a result, MDMX levels increase and bind to MDM2, stabilizing the MDM2/MDMX complex. This complex binds to p53, facilitating its suppression by reducing its transcriptional activity or enhancing its export to the cytoplasm for proteasomal degradation. Consequently, p53 inactivation promotes their survival, proliferation, and resistance to chemotherapy-induced apoptosis. The figure was created in BioRender.com.
期刊介绍:
Oncogenesis is a peer-reviewed open access online journal that publishes full-length papers, reviews, and short communications exploring the molecular basis of cancer and related phenomena. It seeks to promote diverse and integrated areas of molecular biology, cell biology, oncology, and genetics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


