{"title":"Current view on the etiopathogenesis of aplastic anemia.","authors":"Mehmet Ali Ucar, Meryem Sener, Recep Dokuyucu","doi":"10.4196/kjpp.24.214","DOIUrl":null,"url":null,"abstract":"<p><p>Aplastic anemia (AA) is a rare bone marrow failure syndrome marked by hypocellular bone marrow and pancytopenia, typically without abnormal infiltration or reticulin fiber increase. It often presents as acute, severe cytopenia in young adults and can have high mortality if untreated. Recent advancements, including immunosuppressive therapy (IST) combined with eltrombopag and hematopoietic stem cell transplantation (HSCT), have improved patient outcomes. This review discusses current etiopathogenesis involving immune dysregulation, genetic mutations, and environmental triggers. Accurate differential diagnosis, distinguishing AA from myelodysplastic syndromes and paroxysmal nocturnal hemoglobinuria, is essential for effective treatment. We also highlight emerging therapies, such as mismatched unrelated donor (MMUD) transplantation and precision medicine targeting genetic abnormalities. AA, with an incidence of 2-4 per million annually, peaks at ages 15-25 and over 60. These insights continue to reshape AA prognosis and management. This disease typically manifests as acute, severe cytopenia, particularly in young adults, and has a high mortality rate if untreated. Advances in treatment, including IST combined with eltrombopag and HSCT, have significantly improved outcomes. In this review, we explore the current etiopathogenesis, including immune dysregulation, genetic mutations, and environmental factors. The differential diagnosis of AA, distinguishing it from conditions such as myelodysplastic syndromes and paroxysmal nocturnal hemoglobinuria, is critical for tailored treatment. AA remains a rare disease, with an annual incidence of 2-4 per million, and peaks in occurrence during the ages of 15-25 and over 60. These advancements in understanding and managing AA continue to transform its prognosis and patient care.</p>","PeriodicalId":54746,"journal":{"name":"Korean Journal of Physiology & Pharmacology","volume":" ","pages":"399-408"},"PeriodicalIF":2.2000,"publicationDate":"2025-07-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12198452/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Korean Journal of Physiology & Pharmacology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.4196/kjpp.24.214","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/4/28 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"PHARMACOLOGY & PHARMACY","Score":null,"Total":0}
引用次数: 0
Abstract
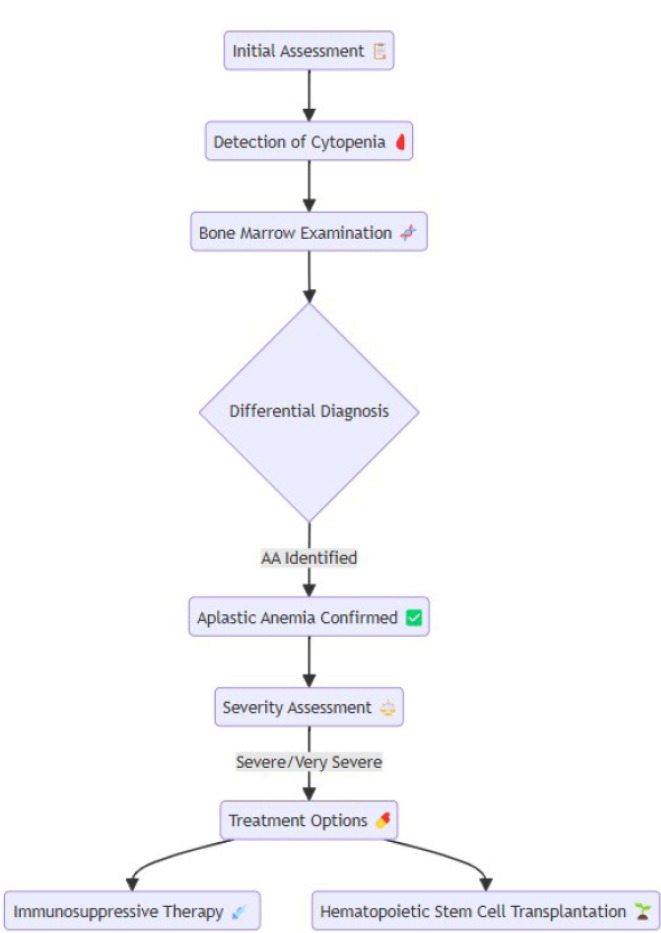
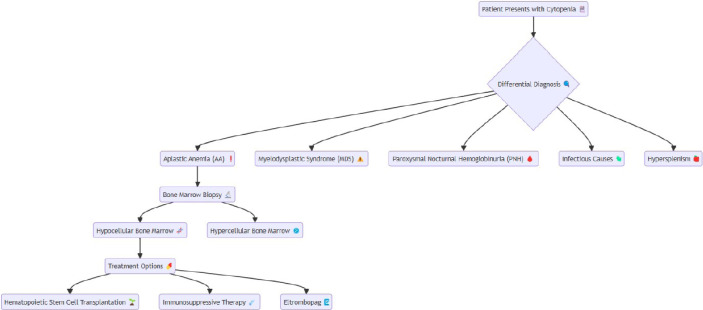
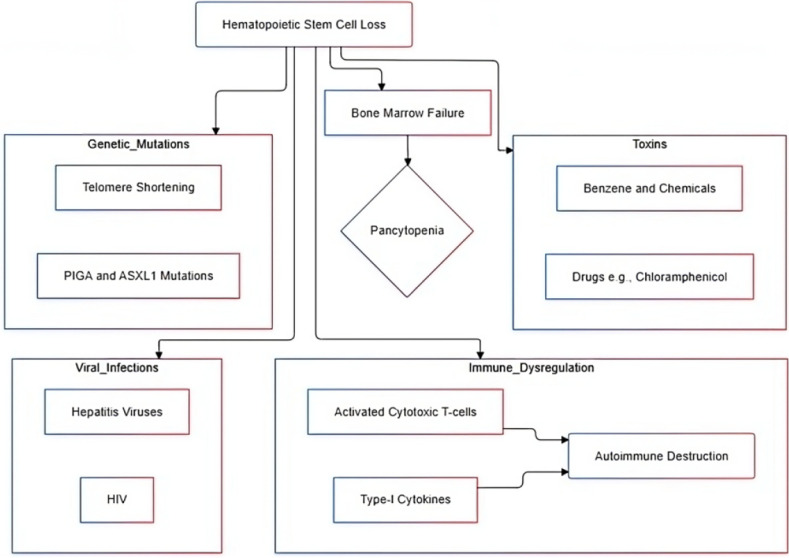
Aplastic anemia (AA) is a rare bone marrow failure syndrome marked by hypocellular bone marrow and pancytopenia, typically without abnormal infiltration or reticulin fiber increase. It often presents as acute, severe cytopenia in young adults and can have high mortality if untreated. Recent advancements, including immunosuppressive therapy (IST) combined with eltrombopag and hematopoietic stem cell transplantation (HSCT), have improved patient outcomes. This review discusses current etiopathogenesis involving immune dysregulation, genetic mutations, and environmental triggers. Accurate differential diagnosis, distinguishing AA from myelodysplastic syndromes and paroxysmal nocturnal hemoglobinuria, is essential for effective treatment. We also highlight emerging therapies, such as mismatched unrelated donor (MMUD) transplantation and precision medicine targeting genetic abnormalities. AA, with an incidence of 2-4 per million annually, peaks at ages 15-25 and over 60. These insights continue to reshape AA prognosis and management. This disease typically manifests as acute, severe cytopenia, particularly in young adults, and has a high mortality rate if untreated. Advances in treatment, including IST combined with eltrombopag and HSCT, have significantly improved outcomes. In this review, we explore the current etiopathogenesis, including immune dysregulation, genetic mutations, and environmental factors. The differential diagnosis of AA, distinguishing it from conditions such as myelodysplastic syndromes and paroxysmal nocturnal hemoglobinuria, is critical for tailored treatment. AA remains a rare disease, with an annual incidence of 2-4 per million, and peaks in occurrence during the ages of 15-25 and over 60. These advancements in understanding and managing AA continue to transform its prognosis and patient care.
期刊介绍:
The Korean Journal of Physiology & Pharmacology (Korean J. Physiol. Pharmacol., KJPP) is the official journal of both the Korean Physiological Society (KPS) and the Korean Society of Pharmacology (KSP). The journal launched in 1997 and is published bi-monthly in English. KJPP publishes original, peer-reviewed, scientific research-based articles that report successful advances in physiology and pharmacology. KJPP welcomes the submission of all original research articles in the field of physiology and pharmacology, especially the new and innovative findings. The scope of researches includes the action mechanism, pharmacological effect, utilization, and interaction of chemicals with biological system as well as the development of new drug targets. Theoretical articles that use computational models for further understanding of the physiological or pharmacological processes are also welcomed. Investigative translational research articles on human disease with an emphasis on physiology or pharmacology are also invited. KJPP does not publish work on the actions of crude biological extracts of either unknown chemical composition (e.g. unpurified and unvalidated) or unknown concentration. Reviews are normally commissioned, but consideration will be given to unsolicited contributions. All papers accepted for publication in KJPP will appear simultaneously in the printed Journal and online.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


