Malin Fromme, Fabienne Klebingat, Paul Ellis, Pavel Strnad
{"title":"Alpha-1 antitrypsin deficiency-associated liver disease: From understudied disorder to the poster child of genetic medicine.","authors":"Malin Fromme, Fabienne Klebingat, Paul Ellis, Pavel Strnad","doi":"10.1097/HC9.0000000000000699","DOIUrl":null,"url":null,"abstract":"<p><p>Alpha-1 antitrypsin deficiency (AATD) constitutes an inborn disorder arising due to mutations in alpha-1 antitrypsin (AAT), a secreted protease inhibitor produced primarily in hepatocytes. It leads to diminished serum AAT levels, and this loss-of-function predisposes to chronic obstructive pulmonary disease and lung emphysema. The characteristic Pi*Z mutation results in hepatic Z-AAT accumulation. In its homozygous form (Pi*ZZ genotype), it is responsible for the majority of severe AATD cases and can cause both pediatric and adult liver disease, while the heterozygous form (Pi*MZ) is considered a disease modifier that becomes apparent primarily in the presence of other comorbidities or risk factors. In the current review, we collate conditions associated with AATD, introduce typical AAT variants, and discuss our understanding of disease pathogenesis. We present both cross-sectional and longitudinal data informing about the natural disease history and noninvasive tools that can be used for disease stratification as well as a basis for disease monitoring. Given that AATD-associated liver disease is highly heterogeneous, we discuss the risk factors affecting disease progression. While the loss-of-function lung disease is treated by weekly intravenous administration of purified AAT, recombinant modified AAT and oral protease inhibitors are currently in clinical trials. Among the liver candidates, small interfering RNA fazirsiran efficiently suppresses AAT production and is currently in phase 3 clinical trial, while several other genetic approaches, such as RNA editing, are at earlier stages. In summary, AATD represents a systemic disorder increasingly seen in the hepatologic routine and requiring thorough interdisciplinary care, since the currently ongoing clinical trials often address only one of the organs it affects.</p>","PeriodicalId":12978,"journal":{"name":"Hepatology Communications","volume":"9 5","pages":""},"PeriodicalIF":5.6000,"publicationDate":"2025-04-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11999460/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hepatology Communications","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1097/HC9.0000000000000699","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/1 0:00:00","PubModel":"eCollection","JCR":"Q1","JCRName":"GASTROENTEROLOGY & HEPATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
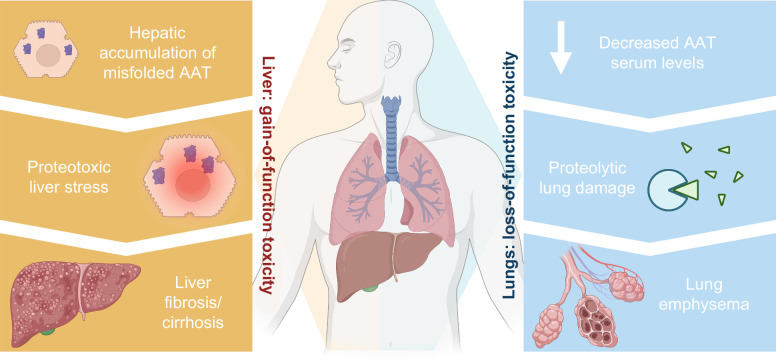
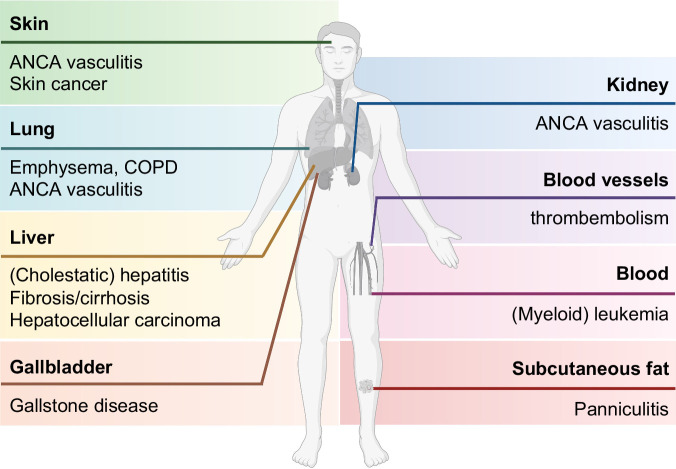
Alpha-1 antitrypsin deficiency (AATD) constitutes an inborn disorder arising due to mutations in alpha-1 antitrypsin (AAT), a secreted protease inhibitor produced primarily in hepatocytes. It leads to diminished serum AAT levels, and this loss-of-function predisposes to chronic obstructive pulmonary disease and lung emphysema. The characteristic Pi*Z mutation results in hepatic Z-AAT accumulation. In its homozygous form (Pi*ZZ genotype), it is responsible for the majority of severe AATD cases and can cause both pediatric and adult liver disease, while the heterozygous form (Pi*MZ) is considered a disease modifier that becomes apparent primarily in the presence of other comorbidities or risk factors. In the current review, we collate conditions associated with AATD, introduce typical AAT variants, and discuss our understanding of disease pathogenesis. We present both cross-sectional and longitudinal data informing about the natural disease history and noninvasive tools that can be used for disease stratification as well as a basis for disease monitoring. Given that AATD-associated liver disease is highly heterogeneous, we discuss the risk factors affecting disease progression. While the loss-of-function lung disease is treated by weekly intravenous administration of purified AAT, recombinant modified AAT and oral protease inhibitors are currently in clinical trials. Among the liver candidates, small interfering RNA fazirsiran efficiently suppresses AAT production and is currently in phase 3 clinical trial, while several other genetic approaches, such as RNA editing, are at earlier stages. In summary, AATD represents a systemic disorder increasingly seen in the hepatologic routine and requiring thorough interdisciplinary care, since the currently ongoing clinical trials often address only one of the organs it affects.
期刊介绍:
Hepatology Communications is a peer-reviewed, online-only, open access journal for fast dissemination of high quality basic, translational, and clinical research in hepatology. Hepatology Communications maintains high standard and rigorous peer review. Because of its open access nature, authors retain the copyright to their works, all articles are immediately available and free to read and share, and it is fully compliant with funder and institutional mandates. The journal is committed to fast publication and author satisfaction.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


