Jonathan Marquez, Lauren O'Sullivan, Audrey E Squire, Ginny L Ryan, Katherine E Debiec, Anne-Marie Amies Oelschlager, Margaret P Adam
{"title":"Case Report: a novel variant in <i>WT1</i> leads to focal segmental glomerulosclerosis and uterovaginal anomalies through exon skipping.","authors":"Jonathan Marquez, Lauren O'Sullivan, Audrey E Squire, Ginny L Ryan, Katherine E Debiec, Anne-Marie Amies Oelschlager, Margaret P Adam","doi":"10.3389/fneph.2025.1542475","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Podocytopathies are a varied set of renal diseases in which podocytes are unable to perform their typical filtration function within the glomerulus. This typically leads to edema, proteinuria, and hypoalbuminemia early in life. Among podocytopathies, focal segmental glomerulosclerosis (FSGS) is characterized by histology demonstrating segmental and focal sclerosis of the glomerular tuft. FSGS affects an estimated 1-20 per one million individuals and leads to significant morbidity and mortality related to renal failure. While FSGS can be attributed to many causes, such as drug reactions and infections, underlying pathogenic genetic variants play an increasingly well-recognized role in this disease.</p><p><strong>Case: </strong>A 38-year-old 46,XX female patient of self-reported Cambodian ancestry was evaluated due to her history of atypical uterovaginal morphology. She had a history of hypertension and nephrotic range proteinuria that was diagnosed early in adulthood. A kidney biopsy at that time revealed FSGS. Following worsening renal function and subsequent end-stage renal disease (ESRD), she underwent a kidney transplant at 33 years of age. After kidney transplant, she presented with hematocolpos and was found to have distal vaginal atresia and an arcuate uterus. She underwent vaginoplasty and then had regular menses. She was noted to have persistently elevated follicle stimulating hormone levels, consistent with primary ovarian insufficiency, but with normal anti-Müllerian hormone levels. Assessment of her family history was suggestive of other individuals in her family with similar renal disease and uterine differences. Genetic analysis identified a <i>WT1</i> variant (c.1338A>C; p. =) of uncertain significance that is also present in her similarly affected mother. To help clarify the potential impact of this variant, we completed a mini-gene assay to detect <i>in vitro</i> splicing changes in the presence of the <i>WT1</i> variant sequence uncovered in this individual. This demonstrated resultant aberrant splicing that further supports the pathogenicity of the uncovered variant for this individual.</p><p><strong>Conclusions: </strong>To our knowledge, this represents the first case of a podocytopathy with co-occurring uterovaginal anomalies due to exon skipping in <i>WT1</i>. The patient exhibited a severe course of chronic kidney dysfunction requiring a kidney transplant. Clinical RNA sequencing to clarify variants impacting splicing remains challenging due to tissue- specific gene expression for genes such as <i>WT1</i>, thus, research-based assays may be beneficial to understand the consequence of rare or previously uncharacterized variants.</p>","PeriodicalId":73091,"journal":{"name":"Frontiers in nephrology","volume":"5 ","pages":"1542475"},"PeriodicalIF":0.0000,"publicationDate":"2025-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11997443/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in nephrology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fneph.2025.1542475","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Podocytopathies are a varied set of renal diseases in which podocytes are unable to perform their typical filtration function within the glomerulus. This typically leads to edema, proteinuria, and hypoalbuminemia early in life. Among podocytopathies, focal segmental glomerulosclerosis (FSGS) is characterized by histology demonstrating segmental and focal sclerosis of the glomerular tuft. FSGS affects an estimated 1-20 per one million individuals and leads to significant morbidity and mortality related to renal failure. While FSGS can be attributed to many causes, such as drug reactions and infections, underlying pathogenic genetic variants play an increasingly well-recognized role in this disease.
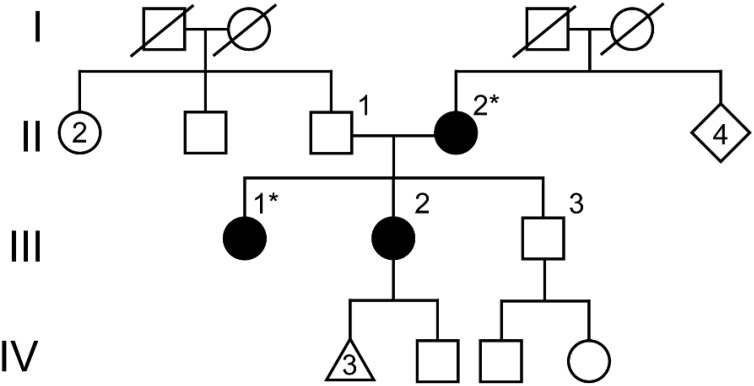
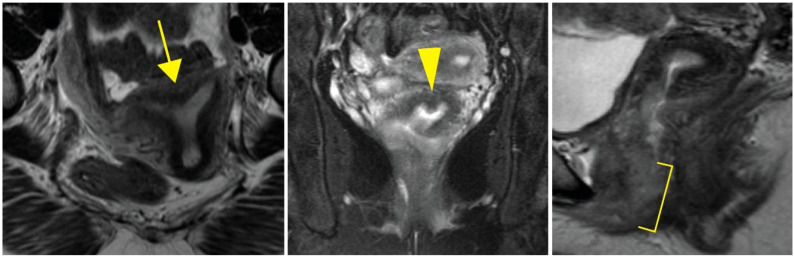
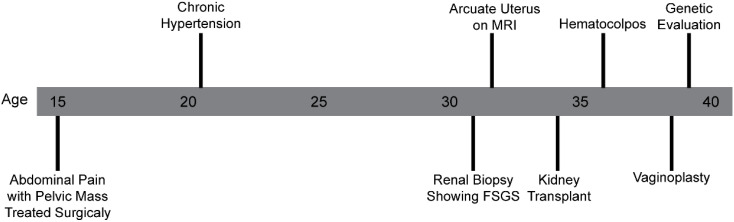
Case: A 38-year-old 46,XX female patient of self-reported Cambodian ancestry was evaluated due to her history of atypical uterovaginal morphology. She had a history of hypertension and nephrotic range proteinuria that was diagnosed early in adulthood. A kidney biopsy at that time revealed FSGS. Following worsening renal function and subsequent end-stage renal disease (ESRD), she underwent a kidney transplant at 33 years of age. After kidney transplant, she presented with hematocolpos and was found to have distal vaginal atresia and an arcuate uterus. She underwent vaginoplasty and then had regular menses. She was noted to have persistently elevated follicle stimulating hormone levels, consistent with primary ovarian insufficiency, but with normal anti-Müllerian hormone levels. Assessment of her family history was suggestive of other individuals in her family with similar renal disease and uterine differences. Genetic analysis identified a WT1 variant (c.1338A>C; p. =) of uncertain significance that is also present in her similarly affected mother. To help clarify the potential impact of this variant, we completed a mini-gene assay to detect in vitro splicing changes in the presence of the WT1 variant sequence uncovered in this individual. This demonstrated resultant aberrant splicing that further supports the pathogenicity of the uncovered variant for this individual.
Conclusions: To our knowledge, this represents the first case of a podocytopathy with co-occurring uterovaginal anomalies due to exon skipping in WT1. The patient exhibited a severe course of chronic kidney dysfunction requiring a kidney transplant. Clinical RNA sequencing to clarify variants impacting splicing remains challenging due to tissue- specific gene expression for genes such as WT1, thus, research-based assays may be beneficial to understand the consequence of rare or previously uncharacterized variants.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


