Elisa Bregant, Elena Betto, Chiara Dal Secco, Jessica Zucco, Federica Baldan, Lorenzo Allegri, Incoronata Renata Lonigro, Flavio Faletra, Lorenzo Verriello, Giuseppe Damante, Catia Mio
{"title":"The molecular landscape of hereditary ataxia: a single-center study.","authors":"Elisa Bregant, Elena Betto, Chiara Dal Secco, Jessica Zucco, Federica Baldan, Lorenzo Allegri, Incoronata Renata Lonigro, Flavio Faletra, Lorenzo Verriello, Giuseppe Damante, Catia Mio","doi":"10.1007/s00439-025-02744-y","DOIUrl":null,"url":null,"abstract":"<p><p>Hereditary ataxia (HA) is a heterogeneous group of complex neurological disorders, which represent a diagnostic challenge due to their diverse phenotypes and genetic etiologies. Next-generation sequencing (NGS) has revolutionized the field of neurogenetics, improving the identification of ataxia-associated genes. Notwithstanding, repeat expansions analysis remains a cornerstone in the diagnostic workflow of these diseases. Here we describe the molecular characterization of a consecutive single-center series of 70 patients with genetically uncharacterized HA. Patients' samples were analyzed for known HA-associated repeat expansions as first tier and negative ones were analyzed by whole exome sequencing (WES) as second tier. Overall, we identified pathogenic/likely pathogenic variants in 40% (n = 28/70) and variants of unknown significance (VUS) in 20% (n = 14/70) of cases. In particular, 10 patients (14.3%, n = 10/70) presented pathogenic repeat expansions while 18 cases (30%, n = 18/60) harbored at least a single nucleotide variant (SNV) or a copy number variant (CNV) in HA or HSP-related genes. WES allowed assessing complex neurological diseases (i.e., leukodystrophies, cerebrotendinous xanthomatosis and atypical xeroderma pigmentosum), which are not usually referred as pure genetic ataxias. Our data suggests that the combined use of repeat expansion analysis and WES, coupled to detailed clinical phenotyping, is able to detect the molecular alteration underpinning ataxia in almost 50% cases, regardless of the hereditary pattern. Indeed, NGS-based tests are fundamental to acknowledge novel HA-associated genes useful to explain the remaining wide fraction of negative tests. Nowadays, this gap is problematic since these patients could not benefit from an etiological diagnosis of their disease that allows prognostic trajectories and prenatal/preimplantation diagnosis.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":"144 5","pages":"545-557"},"PeriodicalIF":3.6000,"publicationDate":"2025-05-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12033174/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-025-02744-y","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/4/10 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
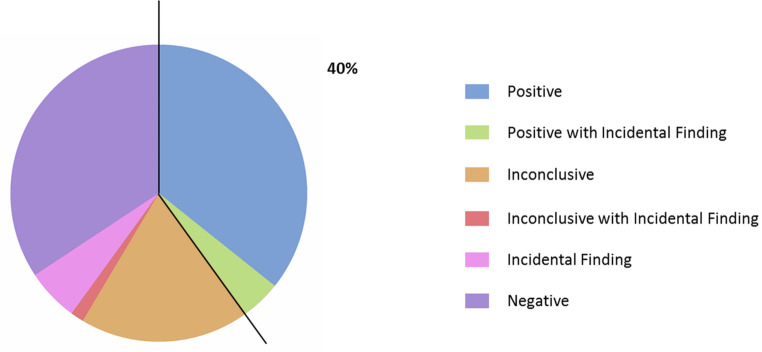
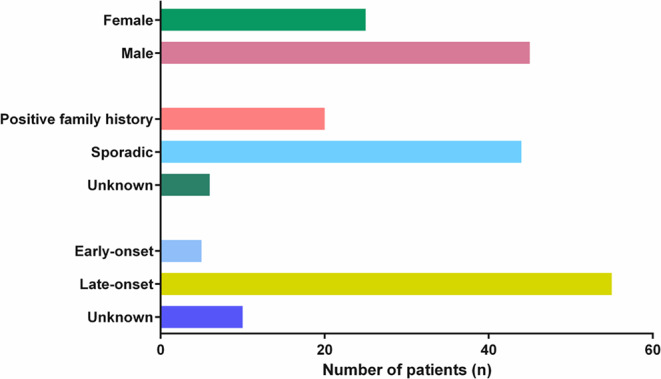
Hereditary ataxia (HA) is a heterogeneous group of complex neurological disorders, which represent a diagnostic challenge due to their diverse phenotypes and genetic etiologies. Next-generation sequencing (NGS) has revolutionized the field of neurogenetics, improving the identification of ataxia-associated genes. Notwithstanding, repeat expansions analysis remains a cornerstone in the diagnostic workflow of these diseases. Here we describe the molecular characterization of a consecutive single-center series of 70 patients with genetically uncharacterized HA. Patients' samples were analyzed for known HA-associated repeat expansions as first tier and negative ones were analyzed by whole exome sequencing (WES) as second tier. Overall, we identified pathogenic/likely pathogenic variants in 40% (n = 28/70) and variants of unknown significance (VUS) in 20% (n = 14/70) of cases. In particular, 10 patients (14.3%, n = 10/70) presented pathogenic repeat expansions while 18 cases (30%, n = 18/60) harbored at least a single nucleotide variant (SNV) or a copy number variant (CNV) in HA or HSP-related genes. WES allowed assessing complex neurological diseases (i.e., leukodystrophies, cerebrotendinous xanthomatosis and atypical xeroderma pigmentosum), which are not usually referred as pure genetic ataxias. Our data suggests that the combined use of repeat expansion analysis and WES, coupled to detailed clinical phenotyping, is able to detect the molecular alteration underpinning ataxia in almost 50% cases, regardless of the hereditary pattern. Indeed, NGS-based tests are fundamental to acknowledge novel HA-associated genes useful to explain the remaining wide fraction of negative tests. Nowadays, this gap is problematic since these patients could not benefit from an etiological diagnosis of their disease that allows prognostic trajectories and prenatal/preimplantation diagnosis.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


