Family mapping of previously identified patients with pathogenic or likely pathogenic ALPL variants using predictive genotyping and detailed phenotyping approach: the FAME case-control study.
Tatiane Vilaca, Fatma Gossiel, Sophie Delaney, Duncan Baker, Sylvia Keigwin, Richard Eastell, Meena Balasubramanian
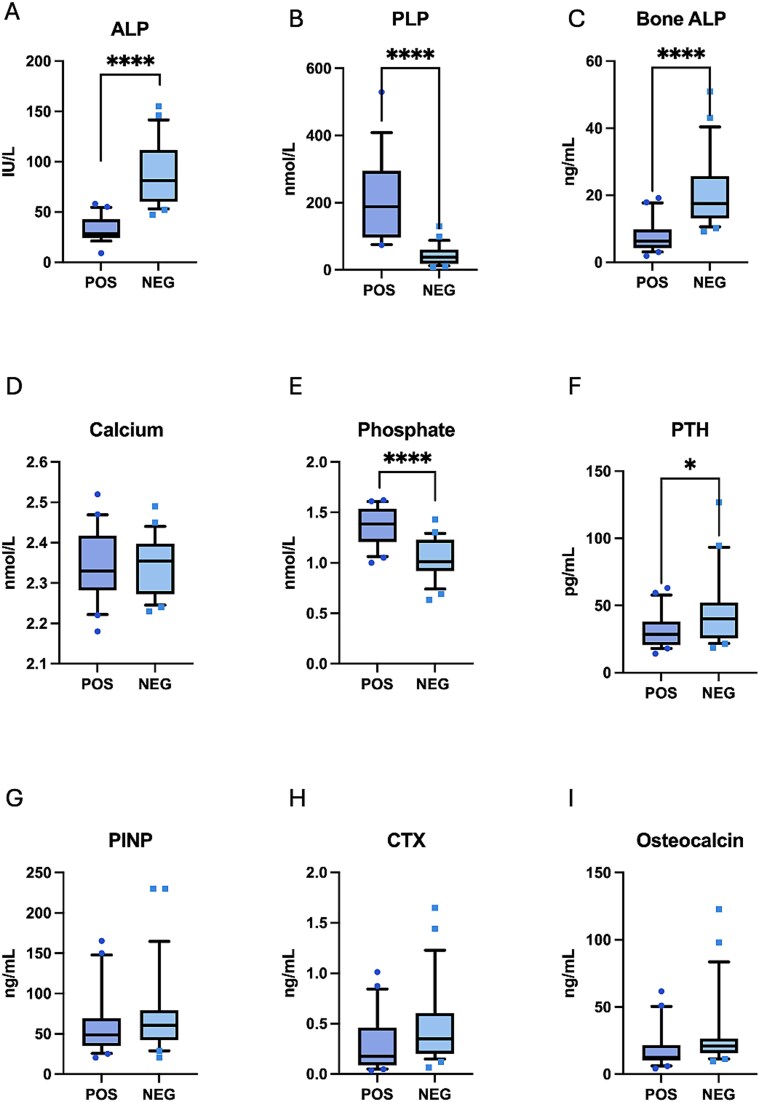
{"title":"Family mapping of previously identified patients with pathogenic or likely pathogenic <i>ALPL</i> variants using predictive genotyping and detailed phenotyping approach: the FAME case-control study.","authors":"Tatiane Vilaca, Fatma Gossiel, Sophie Delaney, Duncan Baker, Sylvia Keigwin, Richard Eastell, Meena Balasubramanian","doi":"10.1093/jbmrpl/ziaf034","DOIUrl":null,"url":null,"abstract":"<p><p>Hypophosphatasia (HPP) is an inborn error of metabolism caused by loss-of-function variants in the <i>ALPL</i> gene, which encodes the tissue nonspecific isozyme of alkaline phosphatase (ALP). There is no typical phenotype in adults. We used a genotyping first approach to determine whether <i>ALPL</i> pathogenic variants were associated with musculoskeletal symptoms, mineral metabolism abnormalities, and an impact on quality of life. We recruited individuals with a pathogenic (or likely pathogenic) variant in <i>ALPL</i> gene (<i>n</i> = 26) and their relatives (<i>n</i> = 44). We performed genetic tests and compared the relatives with positive (<i>n</i> = 20) and negative (<i>n</i> = 24) genetic test. We applied standard questionnaires and physical tests (Brief Pain Inventory [BPI]; Western Ontario and McMaster Universities Arthritis [WOMAC]; Modified Hypophosphatasia Impact Patient Survey; Short Form of 36 Survey [SF-36]; and the Short Physical Performance Battery). In fasting blood samples, we measured creatinine, calcium, phosphate (P), parathyroid hormone (PTH), ALP, bone ALP, 25OHD-, 1,25(OH)2D, CTX, type 1 procollagen N-terminal peptide (PINP), osteocalcin, and tartrate-resistant acid phosphatase5b (TRACP5b). Relatives with positive genetic test had lower ALP (IU/L) [32.5(12.8) vs 87.8(32.6) <i>p</i> < .001], bone ALP (ng/mL) [6.3(4.3, 9.8) vs 17.5 (13.12-25.7) <i>p</i> < .001], PTH (pg/L) [28.6(20.6, 38.1) vs 40.05(25.7, 52.3) <i>p</i> = .03], and higher PLP(nmol/L) [162.0 (91.75, 337.5) vs 37.5 (18.25, 60.5) <i>p</i> < .001] and P(mmol/L) [1.36 (0.18) vs 1.05 (0.2) <i>p</i> < .001]. We did not find significant differences in fractures or musculoskeletal features between the groups. Greater pain scores were observed on BPI in relatives with positive genetic tests, and bone and muscle pain were more often reported by this group, but statistical tests were not significant. No differences were found in physical performance or quality of life. In conclusion, we assessed relatives of individuals with pathogenic or likely pathogenic variants in the <i>ALPL</i> gene regardless of the presence of signs and symptoms. Biochemical abnormalities were more common in gene-positive relatives, but the prevalence of musculoskeletal symptoms was comparable in relatives with positive and negative genetic tests.</p>","PeriodicalId":14611,"journal":{"name":"JBMR Plus","volume":"9 5","pages":"ziaf034"},"PeriodicalIF":2.4000,"publicationDate":"2025-02-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11993272/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JBMR Plus","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/jbmrpl/ziaf034","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/5/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
Hypophosphatasia (HPP) is an inborn error of metabolism caused by loss-of-function variants in the ALPL gene, which encodes the tissue nonspecific isozyme of alkaline phosphatase (ALP). There is no typical phenotype in adults. We used a genotyping first approach to determine whether ALPL pathogenic variants were associated with musculoskeletal symptoms, mineral metabolism abnormalities, and an impact on quality of life. We recruited individuals with a pathogenic (or likely pathogenic) variant in ALPL gene (n = 26) and their relatives (n = 44). We performed genetic tests and compared the relatives with positive (n = 20) and negative (n = 24) genetic test. We applied standard questionnaires and physical tests (Brief Pain Inventory [BPI]; Western Ontario and McMaster Universities Arthritis [WOMAC]; Modified Hypophosphatasia Impact Patient Survey; Short Form of 36 Survey [SF-36]; and the Short Physical Performance Battery). In fasting blood samples, we measured creatinine, calcium, phosphate (P), parathyroid hormone (PTH), ALP, bone ALP, 25OHD-, 1,25(OH)2D, CTX, type 1 procollagen N-terminal peptide (PINP), osteocalcin, and tartrate-resistant acid phosphatase5b (TRACP5b). Relatives with positive genetic test had lower ALP (IU/L) [32.5(12.8) vs 87.8(32.6) p < .001], bone ALP (ng/mL) [6.3(4.3, 9.8) vs 17.5 (13.12-25.7) p < .001], PTH (pg/L) [28.6(20.6, 38.1) vs 40.05(25.7, 52.3) p = .03], and higher PLP(nmol/L) [162.0 (91.75, 337.5) vs 37.5 (18.25, 60.5) p < .001] and P(mmol/L) [1.36 (0.18) vs 1.05 (0.2) p < .001]. We did not find significant differences in fractures or musculoskeletal features between the groups. Greater pain scores were observed on BPI in relatives with positive genetic tests, and bone and muscle pain were more often reported by this group, but statistical tests were not significant. No differences were found in physical performance or quality of life. In conclusion, we assessed relatives of individuals with pathogenic or likely pathogenic variants in the ALPL gene regardless of the presence of signs and symptoms. Biochemical abnormalities were more common in gene-positive relatives, but the prevalence of musculoskeletal symptoms was comparable in relatives with positive and negative genetic tests.
低磷酸酶(HPP)是由ALPL基因的功能缺失变异引起的先天性代谢错误,该基因编码碱性磷酸酶(ALP)的组织非特异性同工酶。在成人中没有典型的表型。我们首先采用基因分型方法来确定ALPL致病变异是否与肌肉骨骼症状、矿物质代谢异常以及对生活质量的影响相关。我们招募了携带致病性(或可能致病性)ALPL基因变异的个体(n = 26)及其亲属(n = 44)。对基因检测阳性(n = 20)和阴性(n = 24)的亲属进行比较。我们采用标准问卷和体格测试(简短疼痛量表[BPI];关节炎[WOMAC];改良磷酸酶减退症影响患者调查36调查简表[SF-36];和短物理性能电池)。在空腹血液样本中,我们测量了肌酐、钙、磷酸盐(P)、甲状旁腺激素(PTH)、ALP、骨ALP、25OHD-、1,25(OH)2D、CTX、1型前胶原n端肽(PINP)、骨钙素和抗酒石酸盐酸性磷酸酶5b (TRACP5b)。基因检测阳性的亲属ALP (IU/L)较低[32.5(12.8)vs 87.8(32.6) p p p =。[03]和更高的PLP(nmol/L) [162.0 (91.75, 337.5) vs 37.5 (18.25, 60.5) p p ALPL基因,无论有无体征和症状。生化异常在基因阳性的亲属中更为常见,但肌肉骨骼症状的患病率在基因检测阳性和阴性的亲属中相当。


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


