Shira Shur, Anna K Sommer, Andrew Latchford, Isabel Spier, Lior H Katz
{"title":"A review of APC somatic mosaicism and specific APC variants - I1307K and promotor variants.","authors":"Shira Shur, Anna K Sommer, Andrew Latchford, Isabel Spier, Lior H Katz","doi":"10.1007/s10689-025-00464-w","DOIUrl":null,"url":null,"abstract":"<p><p>In the majority of patients with a classical Familial Adenomatous Polyposis (FAP) a pathogenic APC germline variant is identified; usually these are truncating variants in the coding region of APC. However, there are some special circumstances in which FAP is not the result of a pathogenic heterozygous germline variant in APC (mosaicism) and tspecific APC variants which do not cause FAP (I1307K and promotor variants). This paper will discuss these three conditions. APC somatic (postzygotic) mosaicism can be identified in up to 50% of unexplained adenomatous polyposis cases. The ability to identify APC postzygotic mosaicism depends on the the detection method (today usually next-generation sequencing) and also the tissue being analysed (investigation of multiple colorectal adenomas is more sensitive than leukocyte DNA). Identifying mosaicism has important implications in terms of an individual's management and managing risk in family members. The I1307K variant in APC is prevalent among Ashkenazi Jews (AJ) but can also be found in Sephardi Jews and individuals of non-Jewish descent. While this variant does not cause polyposis, it increases the risk of colorectal cancer (CRC) by 1.68-fold in AJ individuals. However, the link between the I1307K variant and CRC risk in non-AJ populations, is less well-established. Furthermore, its potential impact on other types of cancer remains unclear. Consequently, the classification of this variant, along with appropriate screening and surveillance recommendations, remains a subject of ongoing debate among leading medical and genetic organizations. Variants in the APC promotor 1B region cause the relatively newly described condition of gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS). It is said to have an isolated gastric phenotype, with neither duodenal, large bowel nor extra-intestinal manifestations. There are many uncertainties regarding this condition, it's penetrance and management. Lack of clinical data and poor understanding of the natural history of the condition remain significant barriers to developing guidelines to manage this condition.</p>","PeriodicalId":12336,"journal":{"name":"Familial Cancer","volume":"24 2","pages":"39"},"PeriodicalIF":2.0000,"publicationDate":"2025-04-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12003607/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Familial Cancer","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10689-025-00464-w","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
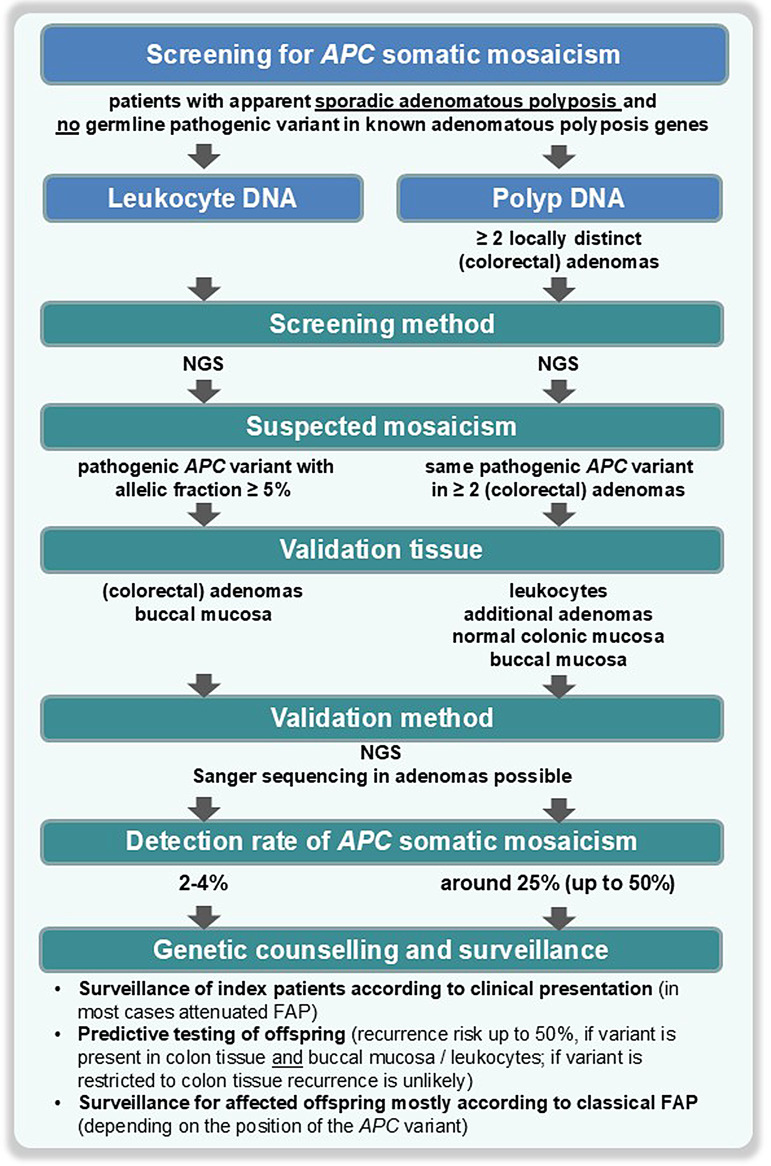
In the majority of patients with a classical Familial Adenomatous Polyposis (FAP) a pathogenic APC germline variant is identified; usually these are truncating variants in the coding region of APC. However, there are some special circumstances in which FAP is not the result of a pathogenic heterozygous germline variant in APC (mosaicism) and tspecific APC variants which do not cause FAP (I1307K and promotor variants). This paper will discuss these three conditions. APC somatic (postzygotic) mosaicism can be identified in up to 50% of unexplained adenomatous polyposis cases. The ability to identify APC postzygotic mosaicism depends on the the detection method (today usually next-generation sequencing) and also the tissue being analysed (investigation of multiple colorectal adenomas is more sensitive than leukocyte DNA). Identifying mosaicism has important implications in terms of an individual's management and managing risk in family members. The I1307K variant in APC is prevalent among Ashkenazi Jews (AJ) but can also be found in Sephardi Jews and individuals of non-Jewish descent. While this variant does not cause polyposis, it increases the risk of colorectal cancer (CRC) by 1.68-fold in AJ individuals. However, the link between the I1307K variant and CRC risk in non-AJ populations, is less well-established. Furthermore, its potential impact on other types of cancer remains unclear. Consequently, the classification of this variant, along with appropriate screening and surveillance recommendations, remains a subject of ongoing debate among leading medical and genetic organizations. Variants in the APC promotor 1B region cause the relatively newly described condition of gastric adenocarcinoma and proximal polyposis of the stomach (GAPPS). It is said to have an isolated gastric phenotype, with neither duodenal, large bowel nor extra-intestinal manifestations. There are many uncertainties regarding this condition, it's penetrance and management. Lack of clinical data and poor understanding of the natural history of the condition remain significant barriers to developing guidelines to manage this condition.
期刊介绍:
In recent years clinical cancer genetics has become increasingly important. Several events, in particular the developments in DNA-based technology, have contributed to this evolution. Clinical cancer genetics has now matured to a medical discipline which is truly multidisciplinary in which clinical and molecular geneticists work together with clinical and medical oncologists as well as with psycho-social workers.
Due to the multidisciplinary nature of clinical cancer genetics most papers are currently being published in a wide variety of journals on epidemiology, oncology and genetics. Familial Cancer provides a forum bringing these topics together focusing on the interests and needs of the clinician.
The journal mainly concentrates on clinical cancer genetics. Most major areas in the field shall be included, such as epidemiology of familial cancer, molecular analysis and diagnosis, clinical expression, treatment and prevention, counselling and the health economics of familial cancer.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


