{"title":"Hemolytic Anemia Due to Gamma-Glutamylcysteine Synthetase Deficiency: A Rare Novel Case in an Arab-Muslim Israeli Child.","authors":"Motti Haimi, Jamal Mahamid","doi":"10.3390/hematolrep17020020","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Gamma-glutamylcysteine synthetase catalyzes the first and rate-limiting step in the synthesis of glutathione. Gamma-glutamylcysteine synthetase deficiency is a very rare condition that has so far been detected so far in nine patients from seven families worldwide. The inheritance of this disorder is autosomal recessive.</p><p><strong>Methods: </strong>We report a case of 4.11-year-old boy, of Arab-Muslim origin, living in an Arab town in Israel who presented at the age of 2 days with severe anemia, reticulocytosis, and leukocytosis. Investigation for common causes of hemolytic anemia was negative (peripheral blood smear was normal, and he had a negative Coombs test, normal G6PD, and normal flow cytometry spherocytosis). The anemia worsened during the following days (hemoglobin (Hb): 7.2 g/dL) and he needed several blood transfusions. NGS (next-generation sequencing) gene panel analysis was performed.</p><p><strong>Results: </strong>In an NGS gene panel analysis for hereditary hemolytic anemias, we found a homozygotic change in the GCLC gene-G53.385.643c379C > T(homo)pArg127Cys-which confirms the diagnosis of gamma-glutamylcysteine synthetase deficiency. An additional rare change was found in this case in the GCLC gene, with unknown clinical significance: g.53373917, c 828 + 3A > G. Except for chronic anemia (Hb levels around 8 g/dL), the child has normal physical and neurological development.</p><p><strong>Conclusions: </strong>This study reports a rare case of gamma-glutamylcysteine synthetase deficiency in a 4.11-year-old Arab-Muslim boy from Israel who presented with severe anemia at 2 days old, aiming to document the first such case in the Middle East and contribute to the medical literature on this extremely rare condition that has only been detected in nine patients worldwide. Genetic analysis revealed a homozygotic change in the GCLC gene, confirming the diagnosis, and while the patient experiences chronic anemia, he maintains normal physical and neurological development, adding valuable insights to the understanding of this rare genetic disorder. An additional rare change was found in this case in the GCLC gene, with unknown clinical significance: g.53373917, c 828 + 3A > G.</p>","PeriodicalId":12829,"journal":{"name":"Hematology Reports","volume":"17 2","pages":""},"PeriodicalIF":1.2000,"publicationDate":"2025-04-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12026703/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Hematology Reports","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/hematolrep17020020","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
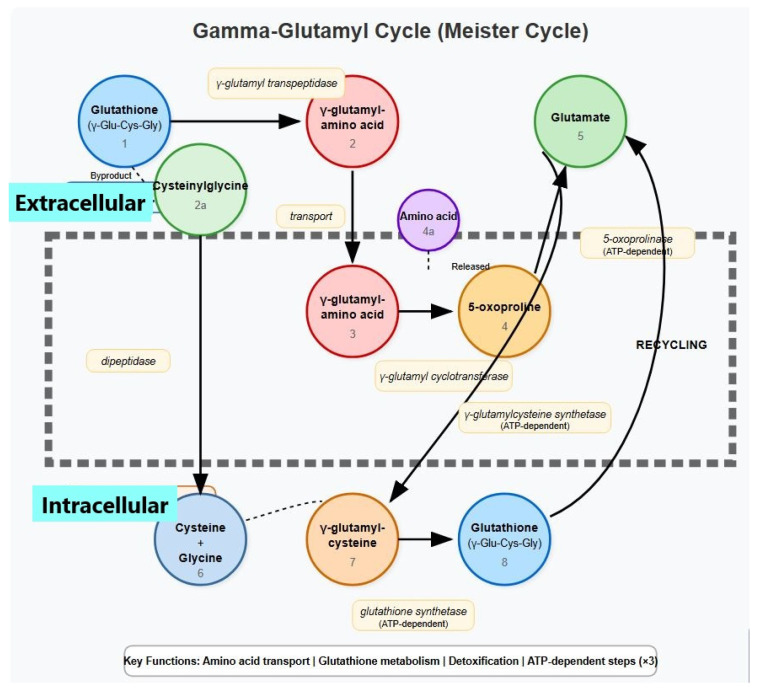
Background: Gamma-glutamylcysteine synthetase catalyzes the first and rate-limiting step in the synthesis of glutathione. Gamma-glutamylcysteine synthetase deficiency is a very rare condition that has so far been detected so far in nine patients from seven families worldwide. The inheritance of this disorder is autosomal recessive.
Methods: We report a case of 4.11-year-old boy, of Arab-Muslim origin, living in an Arab town in Israel who presented at the age of 2 days with severe anemia, reticulocytosis, and leukocytosis. Investigation for common causes of hemolytic anemia was negative (peripheral blood smear was normal, and he had a negative Coombs test, normal G6PD, and normal flow cytometry spherocytosis). The anemia worsened during the following days (hemoglobin (Hb): 7.2 g/dL) and he needed several blood transfusions. NGS (next-generation sequencing) gene panel analysis was performed.
Results: In an NGS gene panel analysis for hereditary hemolytic anemias, we found a homozygotic change in the GCLC gene-G53.385.643c379C > T(homo)pArg127Cys-which confirms the diagnosis of gamma-glutamylcysteine synthetase deficiency. An additional rare change was found in this case in the GCLC gene, with unknown clinical significance: g.53373917, c 828 + 3A > G. Except for chronic anemia (Hb levels around 8 g/dL), the child has normal physical and neurological development.
Conclusions: This study reports a rare case of gamma-glutamylcysteine synthetase deficiency in a 4.11-year-old Arab-Muslim boy from Israel who presented with severe anemia at 2 days old, aiming to document the first such case in the Middle East and contribute to the medical literature on this extremely rare condition that has only been detected in nine patients worldwide. Genetic analysis revealed a homozygotic change in the GCLC gene, confirming the diagnosis, and while the patient experiences chronic anemia, he maintains normal physical and neurological development, adding valuable insights to the understanding of this rare genetic disorder. An additional rare change was found in this case in the GCLC gene, with unknown clinical significance: g.53373917, c 828 + 3A > G.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


