Fatma-Nur Kuzucu, Mustafa Kilic, Abdullah Sezer, Selen Has-Ozhan, Harun Yildiz, Tuba Celen-Yoldas, Fatma-Nese Onat, Melike Uyanik
{"title":"Genotypic and phenotypic characteristics of Turkish patients with phenylalanine metabolism disorders.","authors":"Fatma-Nur Kuzucu, Mustafa Kilic, Abdullah Sezer, Selen Has-Ozhan, Harun Yildiz, Tuba Celen-Yoldas, Fatma-Nese Onat, Melike Uyanik","doi":"10.1007/s11011-025-01582-1","DOIUrl":null,"url":null,"abstract":"<p><p>Phenylketonuria (PKU) is an autosomal recessive disorder of phenylalanine metabolism, in which especially high phenylalanine concentrations cause brain dysfunction. If untreated, this brain dysfunction results in severe intellectual disability, epilepsy, and behavioural problems. We aimed to investigate demographic, clinical, biochemical, and molecular genetic data in patients with phenylalanine metabolism disorder. This study included 99 predominantly Turkish patients diagnosed with phenylalanine metabolism disorder, primarily referred through newborn screening programs. These patients were evaluated at a single center over a 9-year period, from 2013 to 2021. Demographic, clinical, molecular and laboratory data were collected retrospectively. Among the 99 patients, 93 (93.9%) had hyperphenylalaninemia-phenylketonuria, 2 (2.0%) had tetrahydrobiopterin metabolism disorders [one due to 6-pyruvoyl-tetrahydropterin synthase (PTPS) deficiency and the other due to dihydropteridine reductase (DHPR) deficiency], 3 (3.0%) had maternal PKU syndrome (one of whom also had mild phenylketonuria), and 1 (1.0%) had transient hyperphenylalaninemia. The majority of patients belonged to the mild hyperphenylalaninemia-not requiring treatment group. A total of 33 different alleles and 40 genotypes (59.6% compound heterozygous) were identified in the PAH gene, with missense variants accounting for the largest proportion (72.7%). The most frequent PAH gene variants were c.898G > T p.(Ala300Ser) (14.9%), c.1066-11G > A (8.5%), and c.1208C > T p.(Ala403Val) (8.5%), while the most common genotypes were c.898G > T p.(Ala300Ser)/c.898G > T p.(Ala300Ser) (6.4%) and c.898G > T p.(Ala300Ser) /c.1066-11G > A (6.4%), respectively. Among patients with mild hyperphenylalaninemia-not requiring treatment, the predominant genotypes were c.898G > T p.(Ala300Ser)/c.898G > T p.(Ala300Ser) (11.1%), c.898G > T p.(Ala300Ser)/c.1066-11G > A (11.1%), and c.1208C > T p.(Ala403Val)/c.1208C > T p.(Ala403Val) (7.4%), whereas c.842C > T p.(Pro281Leu)/c.842C > T p.(Pro281Leu) (33.3%) was frequently observed in classic PKU patients. The national newborn screening program has significantly improved the prognosis and quality of life for patients through early diagnosis and timely treatment. While the prevalence of hyperphenylalaninemia-phenylketonuria remains high in Turkey, the higher frequency of the hyperphenylalaninemia-not requiring treatment group, compared to European and Asian countries, is considered a favorable outcome. Additionally, the PAH genotype is identified as the primary determinant of the PKU phenotype.</p>","PeriodicalId":18685,"journal":{"name":"Metabolic brain disease","volume":"40 5","pages":"193"},"PeriodicalIF":3.5000,"publicationDate":"2025-04-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12037425/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Metabolic brain disease","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s11011-025-01582-1","RegionNum":3,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
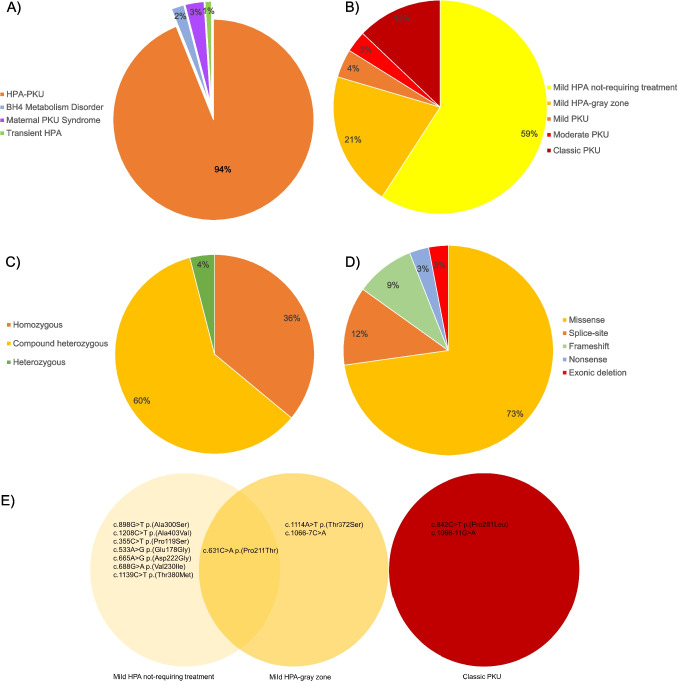
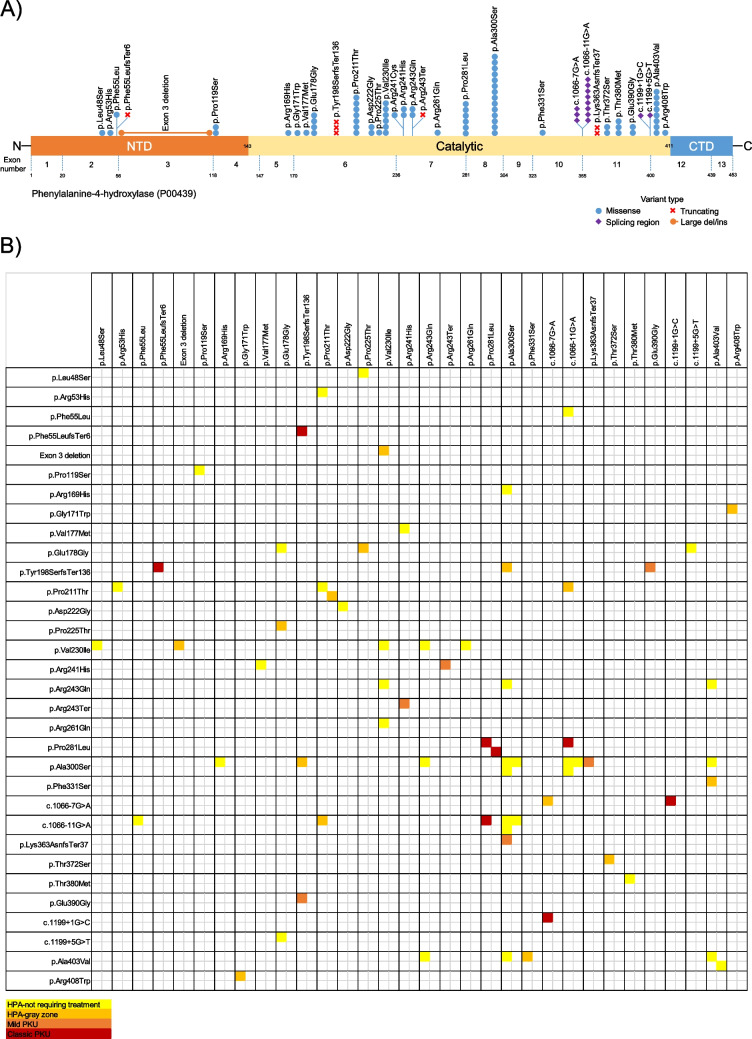
Phenylketonuria (PKU) is an autosomal recessive disorder of phenylalanine metabolism, in which especially high phenylalanine concentrations cause brain dysfunction. If untreated, this brain dysfunction results in severe intellectual disability, epilepsy, and behavioural problems. We aimed to investigate demographic, clinical, biochemical, and molecular genetic data in patients with phenylalanine metabolism disorder. This study included 99 predominantly Turkish patients diagnosed with phenylalanine metabolism disorder, primarily referred through newborn screening programs. These patients were evaluated at a single center over a 9-year period, from 2013 to 2021. Demographic, clinical, molecular and laboratory data were collected retrospectively. Among the 99 patients, 93 (93.9%) had hyperphenylalaninemia-phenylketonuria, 2 (2.0%) had tetrahydrobiopterin metabolism disorders [one due to 6-pyruvoyl-tetrahydropterin synthase (PTPS) deficiency and the other due to dihydropteridine reductase (DHPR) deficiency], 3 (3.0%) had maternal PKU syndrome (one of whom also had mild phenylketonuria), and 1 (1.0%) had transient hyperphenylalaninemia. The majority of patients belonged to the mild hyperphenylalaninemia-not requiring treatment group. A total of 33 different alleles and 40 genotypes (59.6% compound heterozygous) were identified in the PAH gene, with missense variants accounting for the largest proportion (72.7%). The most frequent PAH gene variants were c.898G > T p.(Ala300Ser) (14.9%), c.1066-11G > A (8.5%), and c.1208C > T p.(Ala403Val) (8.5%), while the most common genotypes were c.898G > T p.(Ala300Ser)/c.898G > T p.(Ala300Ser) (6.4%) and c.898G > T p.(Ala300Ser) /c.1066-11G > A (6.4%), respectively. Among patients with mild hyperphenylalaninemia-not requiring treatment, the predominant genotypes were c.898G > T p.(Ala300Ser)/c.898G > T p.(Ala300Ser) (11.1%), c.898G > T p.(Ala300Ser)/c.1066-11G > A (11.1%), and c.1208C > T p.(Ala403Val)/c.1208C > T p.(Ala403Val) (7.4%), whereas c.842C > T p.(Pro281Leu)/c.842C > T p.(Pro281Leu) (33.3%) was frequently observed in classic PKU patients. The national newborn screening program has significantly improved the prognosis and quality of life for patients through early diagnosis and timely treatment. While the prevalence of hyperphenylalaninemia-phenylketonuria remains high in Turkey, the higher frequency of the hyperphenylalaninemia-not requiring treatment group, compared to European and Asian countries, is considered a favorable outcome. Additionally, the PAH genotype is identified as the primary determinant of the PKU phenotype.
苯丙酮尿症(PKU)是一种常染色体隐性苯丙氨酸代谢疾病,特别是高苯丙氨酸浓度可引起脑功能障碍。如果不治疗,这种脑功能障碍会导致严重的智力残疾、癫痫和行为问题。我们的目的是调查苯丙氨酸代谢紊乱患者的人口学、临床、生化和分子遗传学数据。本研究纳入了99名主要为土耳其人的苯丙氨酸代谢紊乱患者,主要通过新生儿筛查项目进行转诊。这些患者在2013年至2021年的9年期间在单一中心进行评估。回顾性收集人口学、临床、分子和实验室资料。99例患者中,高苯丙氨酸血症-苯丙酮尿症93例(93.9%),四氢生物蝶呤代谢紊乱2例(2.0%)[6-丙酮酰四氢蝶呤合成酶(PTPS)缺乏1例,二氢蝶呤还原酶(DHPR)缺乏1例],产妇PKU综合征3例(3.0%)(其中1例伴有轻度苯丙酮尿症),一过性高苯丙氨酸血症1例(1.0%)。大多数患者属于轻度高苯丙氨酸血症-不需要治疗组。PAH基因共鉴定出33个不同的等位基因和40个基因型(59.6%为复合杂合),其中错义变异所占比例最大(72.7%)。最常见的PAH基因变异为c.898 g > T p.(Ala300Ser)(14.9%)、c.1066- 11g > A(8.5%)和c. 1208c > T p.(Ala403Val)(8.5%),而最常见的基因型为c.898 g > T p.(Ala300Ser)/c。898 g > T p。(Ala300Ser)(6.4%)和c.898G > T p。(Ala300Ser) / c。1066-11G > A(6.4%)。在不需要治疗的轻度高苯丙氨酸血症患者中,主要基因型为c.898 g . bbb . 0 T . p.(Ala300Ser)/c。898 g > T p。(Ala300Ser) (11.1%), c.898G > T p。(Ala300Ser) / c。1066 - 11 g >(11.1%),和c.1208C > T p。(Ala403Val) / c。1208C >t p.(Ala403Val)(7.4%),而c. 842c >t p.(Pro281Leu)/c。842C > T p.(Pro281Leu)(33.3%)常见于经典PKU患者。国家新生儿筛查项目通过早期诊断和及时治疗,显著改善了患者的预后和生活质量。虽然高苯丙氨酸血症-苯丙酮尿症在土耳其的患病率仍然很高,但与欧洲和亚洲国家相比,高苯丙氨酸血症-不需要治疗组的发病率较高,这被认为是一个有利的结果。此外,PAH基因型被确定为PKU表型的主要决定因素。
期刊介绍:
Metabolic Brain Disease serves as a forum for the publication of outstanding basic and clinical papers on all metabolic brain disease, including both human and animal studies. The journal publishes papers on the fundamental pathogenesis of these disorders and on related experimental and clinical techniques and methodologies. Metabolic Brain Disease is directed to physicians, neuroscientists, internists, psychiatrists, neurologists, pathologists, and others involved in the research and treatment of a broad range of metabolic brain disorders.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


