Jessica Ortega-Ramos, Mikael Maraschin, Gerardine G. Botte and Joseph A. Gauthier
{"title":"Theoretical determination of a model molecule for the catalytic upcycling of polyethylene and polypropylene†","authors":"Jessica Ortega-Ramos, Mikael Maraschin, Gerardine G. Botte and Joseph A. Gauthier","doi":"10.1039/D4CP04663C","DOIUrl":null,"url":null,"abstract":"<p >Considering the severe environmental and humanitarian implications of global plastic waste accumulation, understanding polyolefin catalytic breakdown is essential. Accordingly, a model compound would improve the reproducibility of experiments and simplify theoretical models. This study aimed to determine the minimum number of monomers necessary to represent the breakdown of polyethylene and polypropylene over metal catalysts. Using density functional theory (DFT) calculations, we evaluated how the polymer's chain length affects reaction energies and energy barriers for C–H and C–C cleavage over stepped transition metal surfaces. We found that chain length does not significantly affect the C–H and C–C cleavage reaction energies and the C–H cleavage energy barriers. Our findings suggest that a small oligomer (less than 10 carbons) could be suitable as a model to study polyethylene's catalytic C–H and C–C cleavage. Although such a simple molecule cannot capture complex transport, entanglement phenomena, and product selectivity observed in full polymers, it may prove useful for determining reaction energetics in complex systems and accelerating catalyst screening.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 21","pages":" 11405-11412"},"PeriodicalIF":2.9000,"publicationDate":"2025-05-14","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp04663c","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
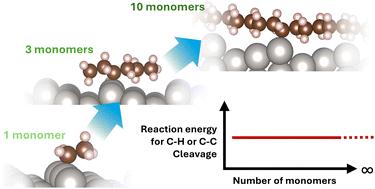
Considering the severe environmental and humanitarian implications of global plastic waste accumulation, understanding polyolefin catalytic breakdown is essential. Accordingly, a model compound would improve the reproducibility of experiments and simplify theoretical models. This study aimed to determine the minimum number of monomers necessary to represent the breakdown of polyethylene and polypropylene over metal catalysts. Using density functional theory (DFT) calculations, we evaluated how the polymer's chain length affects reaction energies and energy barriers for C–H and C–C cleavage over stepped transition metal surfaces. We found that chain length does not significantly affect the C–H and C–C cleavage reaction energies and the C–H cleavage energy barriers. Our findings suggest that a small oligomer (less than 10 carbons) could be suitable as a model to study polyethylene's catalytic C–H and C–C cleavage. Although such a simple molecule cannot capture complex transport, entanglement phenomena, and product selectivity observed in full polymers, it may prove useful for determining reaction energetics in complex systems and accelerating catalyst screening.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


