Binding Energies of Molecules Grafted onto Calcium Surfaces: A Periodic DFT Investigation
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Understanding the spectroscopic signatures of solid-electrolyte interphase (SEI) components is crucial for advancing calcium-based batteries, as the SEI composition directly impacts the performance. X-ray photoelectron spectroscopy (XPS) is a key technique for probing surface chemistry, yet fundamental studies on binding energies of SEI-grafted molecules remain limited. Additionally, accurately computing XPS chemical shifts for surfaces and adsorbates while balancing precision and cost is a challenge. This work employs density functional theory (DFT) with periodic boundary conditions to investigate the adsorption of tetrahydrofuran (THF) and its degradation products on calcium surfaces, including metallic Ca, CaO, and CaH2. A fully hydroxylated CaO surface is also considered. Two primary interaction mechanisms are identified: (i) adsorption via unsaturated carbon atoms and (ii) proton transfer from alcohol groups to CaO. After evaluating various computational approaches, the Slater–Janak scheme is adopted to compute the XPS spectra for O 1s, C 1s, and Ca 2s. Significant shifts in oxygen and carbon binding energies are observed, while these of calcium remain largely unchanged. These findings provide valuable insights into SEI formation and offer useful trends for interpreting XPS spectra, with good agreement observed between simulations and available experimental data.
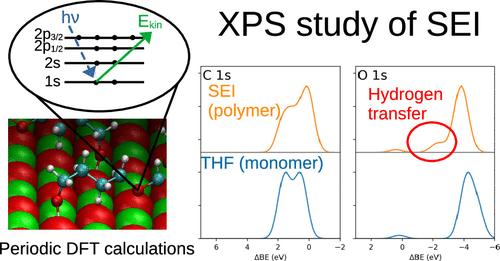
接枝到钙表面的分子结合能:周期性DFT研究
了解固体电解质间相(SEI)成分的光谱特征对于推进钙基电池的发展至关重要,因为SEI成分直接影响电池的性能。x射线光电子能谱(XPS)是探测表面化学的关键技术,但对sei接枝分子结合能的基础研究仍然有限。此外,在平衡精度和成本的同时,准确计算表面和吸附物的XPS化学位移也是一项挑战。本研究采用具有周期性边界条件的密度泛函理论(DFT)研究了四氢呋喃(THF)及其降解产物在钙表面的吸附,包括金属Ca、CaO和CaH2。也考虑了完全羟基化的CaO表面。确定了两种主要的相互作用机制:(i)通过不饱和碳原子吸附和(ii)质子从醇基团转移到CaO。在评估了各种计算方法后,采用Slater-Janak格式计算了O - 1s、C - 1s和Ca - 2s的XPS光谱。氧和碳的结合能发生了显著的变化,而钙的结合能基本保持不变。这些发现为SEI的形成提供了有价值的见解,并为解释XPS光谱提供了有用的趋势,在模拟和现有实验数据之间观察到很好的一致性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


