Insights into the structural and interactional aspects of 1-phenyl-5-(thiophen-2-yl)-1H-tetrazole: crystallographic, Hirshfeld surface, computational, and docking analyses
Akhileshwari P., Preetham R., Sridhar M. A., Sadashiva M. P.
{"title":"Insights into the structural and interactional aspects of 1-phenyl-5-(thiophen-2-yl)-1H-tetrazole: crystallographic, Hirshfeld surface, computational, and docking analyses","authors":"Akhileshwari P., Preetham R., Sridhar M. A., Sadashiva M. P.","doi":"10.1007/s11224-024-02418-x","DOIUrl":null,"url":null,"abstract":"<div><p>Tetrazoles are the bioisoster of carboxylic acid, and their derivatives demonstrated promising anticancer activity. Hybridization of tetrazole moiety with other anticancer pharmacophores may provide novel candidates with anticancer potency. In this regard, the 1-phenyl-5-(thiophen-2-yl)-1H-tetrazole (C<sub>11</sub>H<sub>8</sub>N<sub>4</sub>S) is characterized by LC–MS, C<sup>13</sup> NMR analysis, and single-crystal X-ray diffraction. The molecule crystallizes in the monoclinic crystal system with the space group <i>P</i>2<sub>1</sub>/<i>n</i>. Hirshfeld surface analysis was carried out to quantify the intermolecular interactions and crystal packing. Hirshfeld surface analysis indicates that the most important contributions to the crystal packing are from N–H/H-N (30.7%) interaction. Geometry optimization of the molecule is done using density functional theory (DFT) at the B3LYP hybrid functional. Theoretical structure parameters are compared with the experimentally determined structure. The computed energy gap between the frontier molecular orbitals is 4.81 eV. The RDG calculations revealed the presence of C-H…S and C-H…N noncovalent interaction in the molecule. Molecular docking studies show that the binding affinity between protein and ligand is − 8.03 kcal/mol.</p></div>","PeriodicalId":780,"journal":{"name":"Structural Chemistry","volume":"36 3","pages":"901 - 912"},"PeriodicalIF":2.2000,"publicationDate":"2024-11-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structural Chemistry","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s11224-024-02418-x","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
Tetrazoles are the bioisoster of carboxylic acid, and their derivatives demonstrated promising anticancer activity. Hybridization of tetrazole moiety with other anticancer pharmacophores may provide novel candidates with anticancer potency. In this regard, the 1-phenyl-5-(thiophen-2-yl)-1H-tetrazole (C11H8N4S) is characterized by LC–MS, C13 NMR analysis, and single-crystal X-ray diffraction. The molecule crystallizes in the monoclinic crystal system with the space group P21/n. Hirshfeld surface analysis was carried out to quantify the intermolecular interactions and crystal packing. Hirshfeld surface analysis indicates that the most important contributions to the crystal packing are from N–H/H-N (30.7%) interaction. Geometry optimization of the molecule is done using density functional theory (DFT) at the B3LYP hybrid functional. Theoretical structure parameters are compared with the experimentally determined structure. The computed energy gap between the frontier molecular orbitals is 4.81 eV. The RDG calculations revealed the presence of C-H…S and C-H…N noncovalent interaction in the molecule. Molecular docking studies show that the binding affinity between protein and ligand is − 8.03 kcal/mol.
期刊介绍:
Structural Chemistry is an international forum for the publication of peer-reviewed original research papers that cover the condensed and gaseous states of matter and involve numerous techniques for the determination of structure and energetics, their results, and the conclusions derived from these studies. The journal overcomes the unnatural separation in the current literature among the areas of structure determination, energetics, and applications, as well as builds a bridge to other chemical disciplines. Ist comprehensive coverage encompasses broad discussion of results, observation of relationships among various properties, and the description and application of structure and energy information in all domains of chemistry.
We welcome the broadest range of accounts of research in structural chemistry involving the discussion of methodologies and structures,experimental, theoretical, and computational, and their combinations. We encourage discussions of structural information collected for their chemicaland biological significance.
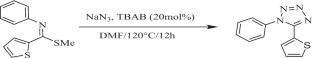

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


