Ruthenium-Catalyzed Enantioselective Alkylation of Sulfenamides: A General Approach for the Synthesis of Drug Relevant S-Methyl and S-Cyclopropyl Sulfoximines
IF 15.6
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
Sulfoximines are increasingly utilized in pharmaceuticals and agrochemicals with all sulfoximine clinical candidates incorporating either an S-methyl or an S-cyclopropyl substituent. Here, we report on a general and efficient sequence for the asymmetric synthesis of both of these sulfoximine substitution patterns. The asymmetric synthesis of sulfilimine intermediates by the first Ru-catalyzed enantioselective alkylation of sulfenamides enables the first examples of enantioselective S-alkylation with monosubstituted diazo compounds. The reaction proceeds at ≤1 mol % Ru-catalyst loading, and for tert-butyl diazoacetate, high yields and ≥98:2 er are achieved for an exceedingly broad range of sulfenamides, including with S-(hetero)aryl, -alkenyl, -methyl, -benzyl, -branched alkyl, and -tert-butyl substituents and for sterically and electronically diverse N-acyl groups. Sulfenamides derived from densely functionalized advanced drug intermediates also alkylated with 99:1 er. After oxidation of an N-pivaloyl S-tert-butyl acetate substituted sulfilimine to the corresponding sulfoximine, treatment with trifluoracetic acid in an aprotic solvent resulted in decarboxylation to the S-methyl N-pivaloyl sulfoximine, while aqueous HCl resulted in both decarboxylation and cleavage of the N-acyl group to give the S-methyl NH sulfoximine. Alternatively, sulfoximine alkylation with dibromoethane followed by acid-mediated decarboxylation provided the S-cyclopropyl sulfoximine. The efficient asymmetric synthesis of the preclinical candidate LTGO-33 and the formal asymmetric synthesis of the phase II clinical candidate ART0380 demonstrate the utility of the disclosed approach.
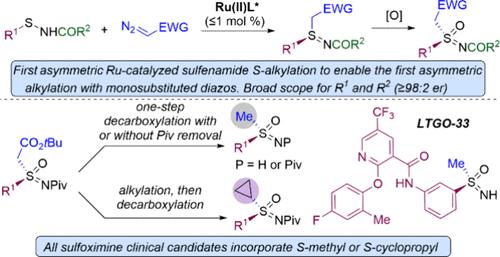
钌催化亚砜胺的对映选择性烷基化:合成与药物相关的s -甲基和s -环丙基亚砜胺的一般方法
亚砜亚胺越来越多地用于制药和农用化学品,所有临床候选亚砜亚胺都含有s -甲基或s -环丙基取代基。在这里,我们报道了不对称合成这两种亚砜亚胺取代图的一般和有效的序列。钌催化的亚砜酰胺的对映选择性烷基化首次合成了亚砜胺中间体,使单取代重氮化合物的对映选择性s烷基化成为第一个例子。该反应在钌催化剂负载≤1mol %的情况下进行,对于重氮乙酸叔丁基,对于非常广泛的亚砜酰胺,包括S-(杂)芳基、-烯基、-甲基、-苄基、-支链烷基和-叔丁基取代基,以及空间和电子上不同的n -酰基,都可以实现高收率和≥98:2 er。由密集功能化的高级药物中间体衍生的磺胺类化合物也与99:1 er烷基化。n -戊酰- s -叔丁基乙酸将亚砜亚胺氧化为相应的亚砜亚胺后,在非质子溶剂中用三氟乙酸处理导致脱羧为s -甲基n -戊酰亚砜亚胺,而水溶液中则导致n -酰基脱羧和裂解,得到s -甲基NH亚砜亚胺。或者,亚砜亚胺与二溴乙烷烷基化,然后酸介导脱羧得到s -环丙基亚砜亚胺。临床前候选药物LTGO-33的高效不对称合成和II期临床候选药物ART0380的正式不对称合成证明了所公开方法的实用性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


