{"title":"Effects of enzyme replacement therapy in sibling cases of hypophosphatasia of varying severities.","authors":"Junko Kanno, Tomohiro Nakagawa, Akinobu Miura, Hirohito Shima, Chisumi Sogi, Miki Kamimura, Ikuma Fujiwara, Kanako Tachikawa, Ryoko Hino, Toshimi Michigami, Atsuo Kikuchi","doi":"10.1297/cpe.2024-0084","DOIUrl":null,"url":null,"abstract":"<p><p>Hypophosphatasia (HPP) is a hereditary disorder characterized by impaired bone mineralization caused by decreased tissue-nonspecific alkaline phosphatase (TNSALP) activity. Specifically, HPP is caused by a loss-of-function variant in the <i>ALPL</i> gene encoding TNSALP. Although genotype-phenotype correlations have been described, phenotypic differences have been reported in patients with the same variants, even within families. The proband, a girl, was suspected to have in utero fractures of the long bones, suggestive of osteogenesis imperfecta. No respiratory impairment was observed after birth; however, the patient's serum alkaline phosphatase level was low. In addition, the patient's perinatal findings were consistent with those of perinatal benign HPP, although the bone symptoms subsequently worsened. The patient's brother, initially suspected to have odonto-HPP due to the premature loss of primary teeth, later developed compression fractures and extraosseous symptoms. Both patients had the same <i>ALPL</i> variants, c. 572A>G(;)1559del, p. Glu191Gly(;)Leu520ArgfsTer86; however, the severity of their conditions differed. Patients with HPP with identical genotypes in the same family may have varying severity levels of HPP. In this case report, both patients received enzyme replacement therapy (ERT), which improved the clinical symptoms. Therefore, for perinatal benign HPP, ERT should be considered if bone symptoms worsen. In addition, odonto-HPP should be closely monitored, and ERT should be considered if bone and extraosseous symptoms arise.</p>","PeriodicalId":10678,"journal":{"name":"Clinical Pediatric Endocrinology","volume":"34 2","pages":"137-143"},"PeriodicalIF":1.2000,"publicationDate":"2025-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11972865/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical Pediatric Endocrinology","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1297/cpe.2024-0084","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/2/13 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
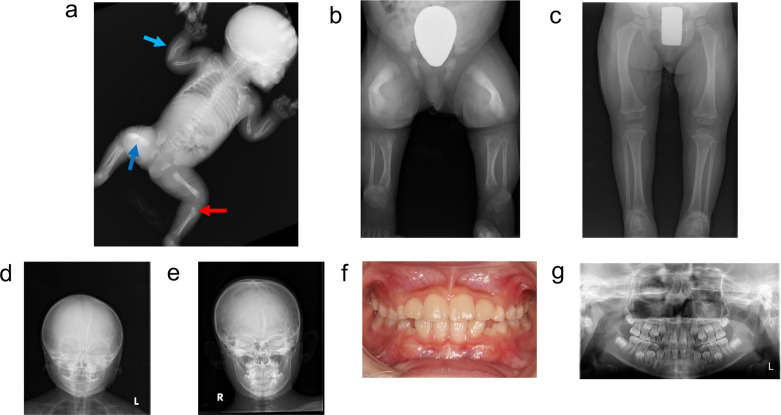
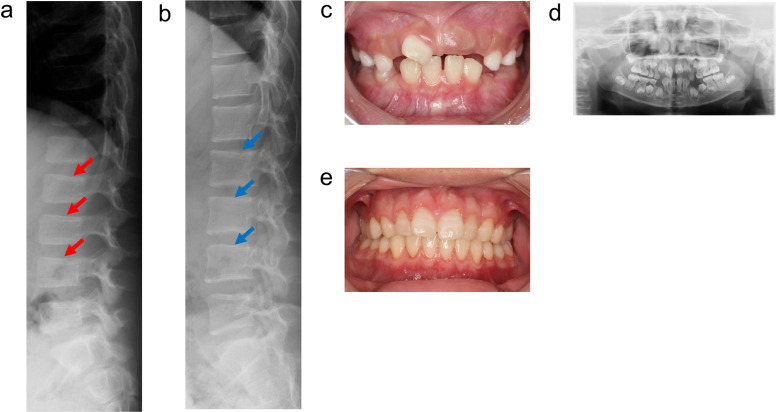
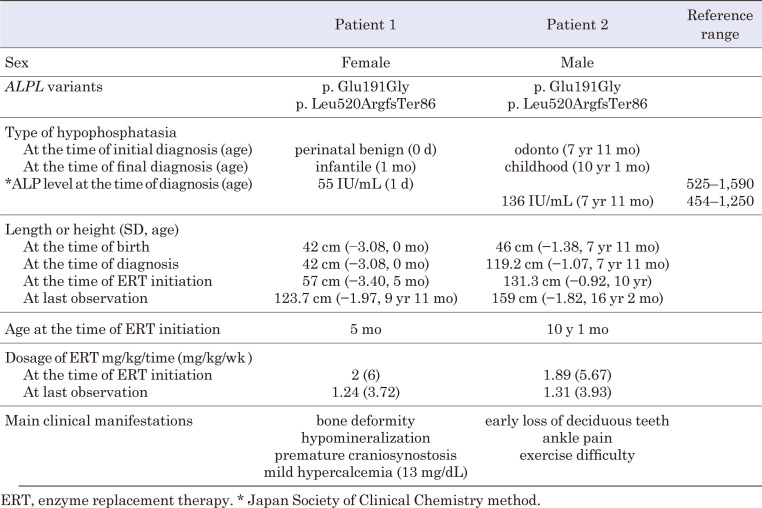
Hypophosphatasia (HPP) is a hereditary disorder characterized by impaired bone mineralization caused by decreased tissue-nonspecific alkaline phosphatase (TNSALP) activity. Specifically, HPP is caused by a loss-of-function variant in the ALPL gene encoding TNSALP. Although genotype-phenotype correlations have been described, phenotypic differences have been reported in patients with the same variants, even within families. The proband, a girl, was suspected to have in utero fractures of the long bones, suggestive of osteogenesis imperfecta. No respiratory impairment was observed after birth; however, the patient's serum alkaline phosphatase level was low. In addition, the patient's perinatal findings were consistent with those of perinatal benign HPP, although the bone symptoms subsequently worsened. The patient's brother, initially suspected to have odonto-HPP due to the premature loss of primary teeth, later developed compression fractures and extraosseous symptoms. Both patients had the same ALPL variants, c. 572A>G(;)1559del, p. Glu191Gly(;)Leu520ArgfsTer86; however, the severity of their conditions differed. Patients with HPP with identical genotypes in the same family may have varying severity levels of HPP. In this case report, both patients received enzyme replacement therapy (ERT), which improved the clinical symptoms. Therefore, for perinatal benign HPP, ERT should be considered if bone symptoms worsen. In addition, odonto-HPP should be closely monitored, and ERT should be considered if bone and extraosseous symptoms arise.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


