Emily Gross, Mark D. Driver, Areesha Saif, Oliver N. Evans and Christopher A. Hunter
{"title":"Solvation energies from atomic surface site interaction points†","authors":"Emily Gross, Mark D. Driver, Areesha Saif, Oliver N. Evans and Christopher A. Hunter","doi":"10.1039/D5CP00635J","DOIUrl":null,"url":null,"abstract":"<p >The surface site interaction model for liquids at equilibrium (SSIMPLE) is a method for calculating thermodynamic properties in a fluid phase based on the use of surface site interaction points (SSIP) to represent all of the non-covalent interactions that molecules make with the environment. Interactions between the SSIPs of two different molecules are governed by a non-polar term and a polar term. Here the formulation originally made for room temperature liquids is generalized to any temperature. We show that the non-polar interaction term is temperature independent while the polar interaction term depends on temperature. This formulation was used to develop a description of the temperature dependence of fluid phase density in terms of an expansion energy, which is based on net intermolecular SSIP interactions. The method is shown to accurately model the temperature dependence of experimentally measured association constants for the formation of 1 : 1 H-bonded complexes in carbon tetrachloride. The atomic interaction point (AIP) version of the SSIP descripiton of 171 different compounds was used in SSIMPLE to calculate room temperature liquid densities that are in good agreement with experimental data. Since non-covalent interactions in the vapour phase can be treated in the same way as liquid phase interactions, SSIMPLE can also be used to calcuate vapour–liquid equilibria (VLE). Experimental VLE data for 196 binary mixtures of 30 miscible compounds was collected, and SSIMPLE was shown to reproduce the experimental behaviour well.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 17","pages":" 8844-8855"},"PeriodicalIF":2.9000,"publicationDate":"2025-04-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00635j?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00635j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
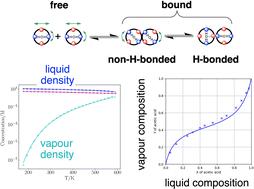
The surface site interaction model for liquids at equilibrium (SSIMPLE) is a method for calculating thermodynamic properties in a fluid phase based on the use of surface site interaction points (SSIP) to represent all of the non-covalent interactions that molecules make with the environment. Interactions between the SSIPs of two different molecules are governed by a non-polar term and a polar term. Here the formulation originally made for room temperature liquids is generalized to any temperature. We show that the non-polar interaction term is temperature independent while the polar interaction term depends on temperature. This formulation was used to develop a description of the temperature dependence of fluid phase density in terms of an expansion energy, which is based on net intermolecular SSIP interactions. The method is shown to accurately model the temperature dependence of experimentally measured association constants for the formation of 1 : 1 H-bonded complexes in carbon tetrachloride. The atomic interaction point (AIP) version of the SSIP descripiton of 171 different compounds was used in SSIMPLE to calculate room temperature liquid densities that are in good agreement with experimental data. Since non-covalent interactions in the vapour phase can be treated in the same way as liquid phase interactions, SSIMPLE can also be used to calcuate vapour–liquid equilibria (VLE). Experimental VLE data for 196 binary mixtures of 30 miscible compounds was collected, and SSIMPLE was shown to reproduce the experimental behaviour well.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


