Sphingosine kinase 2 deficiency impairs VLDL secretion by inhibiting mTORC2 phosphorylation and activating chaperone-mediated autophagy
IF 15.4
1区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
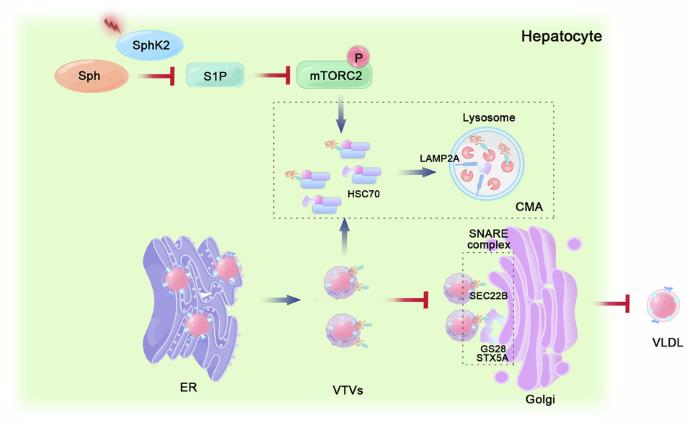
Hepatic very low-density lipoprotein (VLDL) is essential for maintaining lipid metabolism in the liver. Sphingosine kinases (SphKs) are essential rate-limiting enzymes that catalyze sphingosine phosphorylation to Sphingosine-1-phosphate (S1P). SphKs exist as two isoforms, SphK1 and SphK2, both highly expressed in the liver. SphK1 plays a critical role in regulating hepatic inflammation and drug metabolism. This study aimed to determine whether SphK2 regulates hepatic lipid metabolism, particularly VLDL secretion. Immunohistochemical staining revealed decreased SphK2 protein levels within regions proximal to hepatic lipid accumulation in individuals diagnosed with metabolic dysfunction-associated steatotic liver disease (MASLD). Sphk2−/− mice exhibited spontaneous hepatocyte lipid accumulation and reduced VLDL secretion. Proteomic analysis revealed that SphK2 deficiency impaired soluble N-ethylmaleimide-sensitive fusion attachment protein receptor (SNARE) complex interactions involved in vesicular transport and organelle membrane fusion. Furthermore, SphK2 deficiency results in accelerated degradation of the SEC22B, STX5A, and GS28 proteins via chaperone-mediated autophagy (CMA), impeding VLDL transport to the Golgi apparatus. MYH1485, a specific activator of mTOR, induces mTORC2 phosphorylation, thereby inhibiting the degradation of SNARE complexes by CMA and counteracting the lipid accumulation induced by SphK2 deficiency. Exogenous S1P supplementation markedly reversed the reduction in mTORC2 phosphorylation and suppressed CMA, thereby improving VLDL secretion. Our study elucidates an inventive regulatory mechanism by which SphK2 modulates CMA by activating mTORC2 phosphorylation, promoting VLDL secretion, and balancing lipid metabolism in the liver. These findings provide insights into SphK2 function and the underlying mechanisms involved in the regulation of VLDL secretion, which may facilitate MASLD treatment.

鞘氨醇激酶 2 缺乏症通过抑制 mTORC2 磷酸化和激活伴侣介导的自噬作用来损害 VLDL 分泌
肝脏极低密度脂蛋白(VLDL)对维持肝脏脂质代谢至关重要。鞘氨醇激酶(SphKs)是催化鞘氨醇磷酸化生成鞘氨醇-1-磷酸(S1P)的重要限速酶。sphkk以SphK1和SphK2两种亚型存在,均在肝脏中高表达。SphK1在调节肝脏炎症和药物代谢中起关键作用。本研究旨在确定SphK2是否调节肝脏脂质代谢,特别是VLDL分泌。免疫组织化学染色显示,在诊断为代谢功能障碍相关脂肪变性肝病(MASLD)的个体中,肝脏脂质积聚近端区域的SphK2蛋白水平降低。Sphk2−/−小鼠表现出自发的肝细胞脂质积累和VLDL分泌减少。蛋白质组学分析显示,SphK2缺乏损害了参与囊泡运输和细胞器膜融合的可溶性n -乙基马酰亚胺敏感融合附着蛋白受体(SNARE)复合物的相互作用。此外,SphK2缺乏导致SEC22B、STX5A和GS28蛋白通过伴侣介导的自噬(CMA)加速降解,阻碍VLDL转运到高尔基体。MYH1485是mTOR的特异性激活剂,诱导mTORC2磷酸化,从而抑制CMA对SNARE复合物的降解,抵消SphK2缺乏引起的脂质积累。外源性S1P补充显著逆转了mTORC2磷酸化的减少,抑制了CMA,从而改善了VLDL的分泌。我们的研究阐明了SphK2通过激活mTORC2磷酸化、促进VLDL分泌和平衡肝脏脂质代谢来调节CMA的一种创新的调节机制。这些发现为SphK2的功能和VLDL分泌调节的潜在机制提供了见解,这可能有助于MASLD的治疗。SphK2在肝脏VLDL分泌中的作用模型。在肝细胞中,抑制SphK2活性会降低S1P的产生,从而下调mTORC2通路。这一过程通过CMA加速SNARE复合物组分STX5A、GS28和SEC22B的降解,从而调节vtv和高尔基体之间的相互识别,最终减少肝细胞中VLDL的分泌。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Cell Death and Differentiation
生物-生化与分子生物学
CiteScore
24.70
自引率
1.60%
发文量
181
审稿时长
3 months
期刊介绍:
Mission, vision and values of Cell Death & Differentiation:
To devote itself to scientific excellence in the field of cell biology, molecular biology, and biochemistry of cell death and disease.
To provide a unified forum for scientists and clinical researchers
It is committed to the rapid publication of high quality original papers relating to these subjects, together with topical, usually solicited, reviews, meeting reports, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


