B-Power: Investigating the Upper Limits of Enhanced Surface Reactivity in Doped MoTe2
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Transition-metal dichalcogenide (TMD) monolayers have emerged as promising materials in electronics, gas sensing, and electrocatalysis. However, the limited chemical reactivity of TMD basal planes constrains their performance due to the lack of coordinatively unsaturated surface sites. Single-atom doping has shown potential to enhance TMD reactivity, though optimal doping strategies remain uncertain due to an incomplete understanding of the mechanisms and selectivity in reactivity enhancement. This study addresses this gap by investigating boron-doped MoTe2 (B-MoTe2)-the most active candidate identified in prior evaluations of 22 p-block elements. Using density functional theory (DFT) methods, we studied the adsorption behavior of 9 probe molecules: N2O, NO2, NO, N2, CO2, CO, O2, H2O, and H2. Four distinct adsorption behaviors were identified: (i) dissociation of NO2, O2, and H2, (ii) substitution of B by N for NO, (iii) chemisorption of N2O, CO, and H2O, and (iv) physisorption of N2 and CO2. These interactions are driven by the bonding affinity with boron and geometrical constraints of the sheet. The enhanced reactivity of B-MoTe2 arises from boron adopting more favorable orbital hybridization during molecule adsorption, yielding adsorption strengths tens of times higher than those in pristine MoTe2, where interactions are dominated by van der Waals forces. The unique physicochemical properties of B-MoTe2 highlight the potential for tailored reactivity in catalysis and sensing applications and provide a foundational framework for developing effective doping strategies in TMD-based materials.
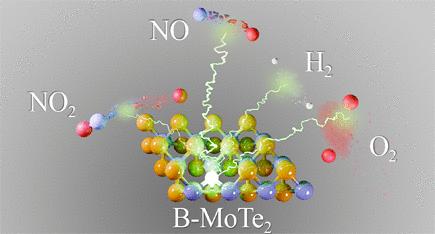
b -功率:研究掺杂MoTe2增强表面反应性的上限
过渡金属二硫化物(TMD)单层膜在电子、气敏和电催化等领域具有广阔的应用前景。然而,由于缺乏配位不饱和表面位点,TMD基面化学反应活性有限,限制了其性能。单原子掺杂显示出增强TMD反应性的潜力,尽管由于对反应性增强的机制和选择性的了解不完全,最佳掺杂策略仍然不确定。本研究通过研究硼掺杂MoTe2 (B-MoTe2)来解决这一空白,硼掺杂MoTe2是在22个p块元素的先前评估中确定的最活跃的候选元素。利用密度泛函理论(DFT)方法,研究了N2O、NO2、NO、N2、CO2、CO、O2、H2O和H2 9种探针分子的吸附行为。发现了四种不同的吸附行为:(i) NO2、O2和H2的解离,(ii) B被N取代NO, (iii) N2O、CO和H2O的化学吸附,以及(iv) N2和CO2的物理吸附。这些相互作用是由与硼的键亲和和薄片的几何约束驱动的。B-MoTe2的反应性增强是由于硼在分子吸附过程中采用了更有利的轨道杂化,其吸附强度比原始MoTe2高数十倍,原始MoTe2的相互作用主要是范德华力。B-MoTe2独特的物理化学性质突出了在催化和传感应用中定制反应性的潜力,并为开发有效的tmd基材料掺杂策略提供了基础框架。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


