Johannes Schörghuber, Nina Bučková, Esther Heid and Georg K. H. Madsen
{"title":"From flat to stepped: active learning frameworks for investigating local structure at copper–water interfaces","authors":"Johannes Schörghuber, Nina Bučková, Esther Heid and Georg K. H. Madsen","doi":"10.1039/D5CP00396B","DOIUrl":null,"url":null,"abstract":"<p >Understanding processes at solid–liquid interfaces at the atomic level is important for applications such as electrocatalysis. Here we explore the effects of different step densities on the structure of interfacial water at the copper–water interface. Utilizing spatially resolved uncertainties, we develop an active learning framework and train a machine-learning force field (MLFF) based on dispersion-corrected density functional theory data. Using molecular dynamics simulations, we investigate structural properties of water molecules in the contact layer, including density profiles, angular distributions, and 2D pair correlation functions. In accordance with previous studies, we observe the formation of two sublayers within the contact layer at the Cu(111) surface, whereas the structure on surfaces with a high step density is dominated by the undercoordinated ridge atoms. By systematically decreasing the step density, we identify the cross-over to when the behavior observed at the flat surface can be locally recovered. Using dimensionality reduction, we identify four distinct types of Cu environments at the interfaces, providing insights into analyzing less idealized surfaces with MLFFs.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 17","pages":" 9169-9177"},"PeriodicalIF":2.9000,"publicationDate":"2025-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2025/cp/d5cp00396b?page=search","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d5cp00396b","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
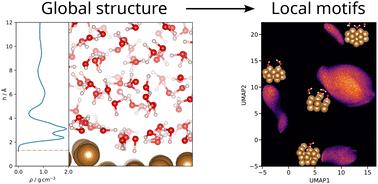
Understanding processes at solid–liquid interfaces at the atomic level is important for applications such as electrocatalysis. Here we explore the effects of different step densities on the structure of interfacial water at the copper–water interface. Utilizing spatially resolved uncertainties, we develop an active learning framework and train a machine-learning force field (MLFF) based on dispersion-corrected density functional theory data. Using molecular dynamics simulations, we investigate structural properties of water molecules in the contact layer, including density profiles, angular distributions, and 2D pair correlation functions. In accordance with previous studies, we observe the formation of two sublayers within the contact layer at the Cu(111) surface, whereas the structure on surfaces with a high step density is dominated by the undercoordinated ridge atoms. By systematically decreasing the step density, we identify the cross-over to when the behavior observed at the flat surface can be locally recovered. Using dimensionality reduction, we identify four distinct types of Cu environments at the interfaces, providing insights into analyzing less idealized surfaces with MLFFs.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


