{"title":"Clinical and Molecular Features of Patients With Congenital Disorders of Glycosylation in Japan","authors":"Nobuhiko Okamoto, Machiko Kadoya, Yoshinao Wada","doi":"10.1002/jmd2.70011","DOIUrl":null,"url":null,"abstract":"<p>Congenital disorders of glycosylation (CDG) are a heterogeneous group of diseases caused by defects in various steps of the glycosylation pathway. There are over 200 known human glycosylation-related disorders. Many of these defects lead to multisystemic manifestations, commonly involving the central nervous system, with symptoms ranging from mild to severe. The phenotypic presentation of CDG can vary significantly. Identifying altered protein glycosylation is crucial for accurate diagnosis. Our research institute has contributed to the CDG diagnostic support center in Japan, developing new analytical techniques utilizing mass spectrometry. These techniques allow for the identification of defects in <i>N</i>-glycosylation, <i>O</i>-glycosylation, and combined glycosylation pathways. Advances in genetic analysis, including whole exome sequencing, have revealed that certain types of CDG are more prevalent than previously recognized. We have contributed to the molecular diagnosis of 66 CDG patients in Japan. Although PMM2-CDG is the most common form of CDG, it was only detected in 17% of the patients in the present study, suggesting that its incidence is much lower in Japan compared to European countries. We also conducted a comprehensive review of case reports of CDG in Japan, further describing the clinical and molecular spectrum of the disease in this population.</p>","PeriodicalId":14930,"journal":{"name":"JIMD reports","volume":"66 3","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2025-04-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jmd2.70011","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JIMD reports","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jmd2.70011","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"Biochemistry, Genetics and Molecular Biology","Score":null,"Total":0}
引用次数: 0
Abstract
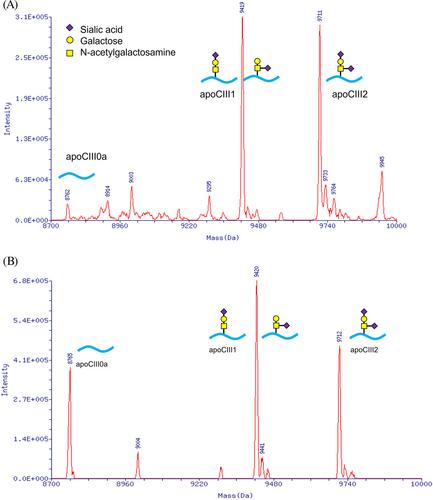
Congenital disorders of glycosylation (CDG) are a heterogeneous group of diseases caused by defects in various steps of the glycosylation pathway. There are over 200 known human glycosylation-related disorders. Many of these defects lead to multisystemic manifestations, commonly involving the central nervous system, with symptoms ranging from mild to severe. The phenotypic presentation of CDG can vary significantly. Identifying altered protein glycosylation is crucial for accurate diagnosis. Our research institute has contributed to the CDG diagnostic support center in Japan, developing new analytical techniques utilizing mass spectrometry. These techniques allow for the identification of defects in N-glycosylation, O-glycosylation, and combined glycosylation pathways. Advances in genetic analysis, including whole exome sequencing, have revealed that certain types of CDG are more prevalent than previously recognized. We have contributed to the molecular diagnosis of 66 CDG patients in Japan. Although PMM2-CDG is the most common form of CDG, it was only detected in 17% of the patients in the present study, suggesting that its incidence is much lower in Japan compared to European countries. We also conducted a comprehensive review of case reports of CDG in Japan, further describing the clinical and molecular spectrum of the disease in this population.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


