Megan A Wallace, Michelle Wille, Jemma Geoghegan, Ryan M Imrie, Edward C Holmes, Xavier A Harrison, Ben Longdon
{"title":"Making sense of the virome in light of evolution and ecology.","authors":"Megan A Wallace, Michelle Wille, Jemma Geoghegan, Ryan M Imrie, Edward C Holmes, Xavier A Harrison, Ben Longdon","doi":"10.1098/rspb.2025.0389","DOIUrl":null,"url":null,"abstract":"<p><p>Understanding the patterns and drivers of viral prevalence and abundance is of key importance for understanding pathogen emergence. Over the last decade, metagenomic sequencing has exponentially expanded our knowledge of the diversity and evolution of viruses associated with all domains of life. However, as most of these 'virome' studies are primarily descriptive, our understanding of the predictors of virus prevalence, abundance and diversity, and their variation in space and time, remains limited. For example, we do not yet understand the relative importance of ecological predictors (e.g. seasonality and habitat) versus evolutionary predictors (e.g. host and virus phylogenies) in driving virus prevalence and diversity. Few studies are set up to reveal the factors that predict the virome composition of individual hosts, populations or species. In addition, most studies of virus ecology represent a snapshot of single species viromes at a single point in time and space. Fortunately, recent studies have begun to use metagenomic data to directly test hypotheses about the evolutionary and ecological factors which drive virus prevalence, sharing and diversity. By synthesizing evidence across studies, we present some over-arching ecological and evolutionary patterns in virome composition, and illustrate the need for additional work to quantify the drivers of virus prevalence and diversity.</p>","PeriodicalId":20589,"journal":{"name":"Proceedings of the Royal Society B: Biological Sciences","volume":"292 2044","pages":"20250389"},"PeriodicalIF":3.5000,"publicationDate":"2025-04-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11961256/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proceedings of the Royal Society B: Biological Sciences","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1098/rspb.2025.0389","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/4/2 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
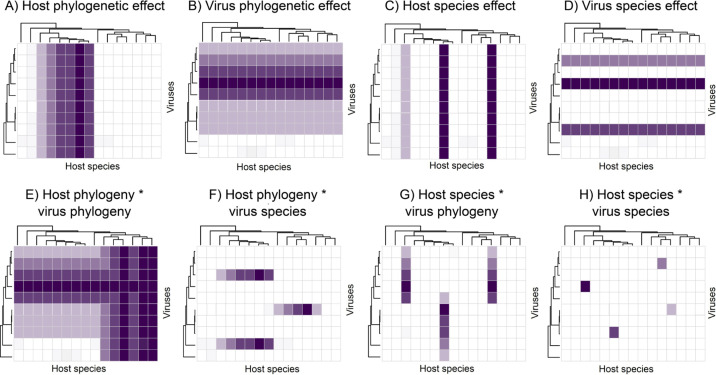
Understanding the patterns and drivers of viral prevalence and abundance is of key importance for understanding pathogen emergence. Over the last decade, metagenomic sequencing has exponentially expanded our knowledge of the diversity and evolution of viruses associated with all domains of life. However, as most of these 'virome' studies are primarily descriptive, our understanding of the predictors of virus prevalence, abundance and diversity, and their variation in space and time, remains limited. For example, we do not yet understand the relative importance of ecological predictors (e.g. seasonality and habitat) versus evolutionary predictors (e.g. host and virus phylogenies) in driving virus prevalence and diversity. Few studies are set up to reveal the factors that predict the virome composition of individual hosts, populations or species. In addition, most studies of virus ecology represent a snapshot of single species viromes at a single point in time and space. Fortunately, recent studies have begun to use metagenomic data to directly test hypotheses about the evolutionary and ecological factors which drive virus prevalence, sharing and diversity. By synthesizing evidence across studies, we present some over-arching ecological and evolutionary patterns in virome composition, and illustrate the need for additional work to quantify the drivers of virus prevalence and diversity.
期刊介绍:
Proceedings B is the Royal Society’s flagship biological research journal, accepting original articles and reviews of outstanding scientific importance and broad general interest. The main criteria for acceptance are that a study is novel, and has general significance to biologists. Articles published cover a wide range of areas within the biological sciences, many have relevance to organisms and the environments in which they live. The scope includes, but is not limited to, ecology, evolution, behavior, health and disease epidemiology, neuroscience and cognition, behavioral genetics, development, biomechanics, paleontology, comparative biology, molecular ecology and evolution, and global change biology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


