Srishti Gupta*, Ajin Rajan, Edvin Fako, Tiago J. Goncalves, Imke B. Müller, Jithin John Varghese, Ansgar Schäfer and Sandip De*,
{"title":"Exploring the Intricacies of Glycerol Hydrodeoxygenation on Copper Surface: A Comprehensive Investigation with the Aid of Machine Learning Force Field","authors":"Srishti Gupta*, Ajin Rajan, Edvin Fako, Tiago J. Goncalves, Imke B. Müller, Jithin John Varghese, Ansgar Schäfer and Sandip De*, ","doi":"10.1021/acs.jpcc.5c0030210.1021/acs.jpcc.5c00302","DOIUrl":null,"url":null,"abstract":"<p >The utilization of biomass for feedstock chemicals often relies on transforming hydroxyl-containing molecules. One such example is glycerol, which can undergo a selective hydrodeoxygenation reaction to produce propanediol, a valuable chemical precursor. Hence, glycerol’s hydrodeoxygenation reaction combines immediate industrial application with the foundation of fundamental research into the reaction class relevant for sustainable feedstock. Given the complex nature of large organic molecules, most modeling work in heterogeneous catalysis focuses on the reactivity of small (C1–2) organics exclusively. Glycerol, characterized by its C3-backbone, has 75 distinct gas-phase conformers with energy variation up to 0.1 eV (<contrib-group><span>Callam, C. S.</span></contrib-group> <cite><i>J. Am. Chem. Soc.</i></cite> <span>2001</span>, <em>123</em>, 11743–11754). When considering its 11 reactive bonds (C–O, C–H, and O–H), the modeling of glycerol’s reactivity spans an extensive conformational and reactive space. High computational costs of density functional theory simulations restrict exhaustive exploration of the factorial reaction space, leading to limited insights into the hydrodeoxygenation (HDO) mechanism and hindering rational catalyst design. Therefore, to date, there is no systematic study focusing on comprehensively sampling the energetics of surface conformers of glycerol and their reactivity. In this study, we employ a message-passing graph neural network architecture (MACE) to develop a machine-learned force-field (MLFF) potential, incorporating active learning to investigate the impact of conformational complexity on the reaction network of glycerol HDO on a Cu(111) surface. Following five iterations, our trained MLFF model accurately predicts surface bound structures with a root-mean-square accuracy of 0.04 eV (<0.6 meV/atom total energy), essential to accurately determine conformational minima of 24 metastable and 26 intermediate states along seven competitive pathways. Conformational sampling uncovers the intricate nature of the complex energy landscape, where conformers with multiple shallow minima lead to nontrivial trends in the transition state energies connecting them. Notably, the investigations predict lower activation barriers for O–H bond scissions of glycerol structures with α- and γ-backbone as compared to β-backbone. This is significant in the case of scission of secondary O–H glycerol bonds where the activation barrier varies up to 0.44 eV depending upon the initial glycerol structure motif. Altogether, we identify dehydrogenation–dehydration–hydrogenation as the dominant pathway resulting in PDO formation on the Cu(111) surface. The selectivity of glyceraldehyde toward C–H bond scission over C–OH bond scission explains the higher selectivity of 1,2-PDO over 1,3-PDO.</p>","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"129 13","pages":"6262–6274 6262–6274"},"PeriodicalIF":3.2000,"publicationDate":"2025-03-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcc.5c00302","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
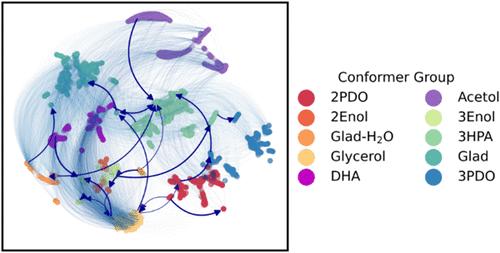
The utilization of biomass for feedstock chemicals often relies on transforming hydroxyl-containing molecules. One such example is glycerol, which can undergo a selective hydrodeoxygenation reaction to produce propanediol, a valuable chemical precursor. Hence, glycerol’s hydrodeoxygenation reaction combines immediate industrial application with the foundation of fundamental research into the reaction class relevant for sustainable feedstock. Given the complex nature of large organic molecules, most modeling work in heterogeneous catalysis focuses on the reactivity of small (C1–2) organics exclusively. Glycerol, characterized by its C3-backbone, has 75 distinct gas-phase conformers with energy variation up to 0.1 eV (Callam, C. S.J. Am. Chem. Soc.2001, 123, 11743–11754). When considering its 11 reactive bonds (C–O, C–H, and O–H), the modeling of glycerol’s reactivity spans an extensive conformational and reactive space. High computational costs of density functional theory simulations restrict exhaustive exploration of the factorial reaction space, leading to limited insights into the hydrodeoxygenation (HDO) mechanism and hindering rational catalyst design. Therefore, to date, there is no systematic study focusing on comprehensively sampling the energetics of surface conformers of glycerol and their reactivity. In this study, we employ a message-passing graph neural network architecture (MACE) to develop a machine-learned force-field (MLFF) potential, incorporating active learning to investigate the impact of conformational complexity on the reaction network of glycerol HDO on a Cu(111) surface. Following five iterations, our trained MLFF model accurately predicts surface bound structures with a root-mean-square accuracy of 0.04 eV (<0.6 meV/atom total energy), essential to accurately determine conformational minima of 24 metastable and 26 intermediate states along seven competitive pathways. Conformational sampling uncovers the intricate nature of the complex energy landscape, where conformers with multiple shallow minima lead to nontrivial trends in the transition state energies connecting them. Notably, the investigations predict lower activation barriers for O–H bond scissions of glycerol structures with α- and γ-backbone as compared to β-backbone. This is significant in the case of scission of secondary O–H glycerol bonds where the activation barrier varies up to 0.44 eV depending upon the initial glycerol structure motif. Altogether, we identify dehydrogenation–dehydration–hydrogenation as the dominant pathway resulting in PDO formation on the Cu(111) surface. The selectivity of glyceraldehyde toward C–H bond scission over C–OH bond scission explains the higher selectivity of 1,2-PDO over 1,3-PDO.
生物质作为原料化学品的利用往往依赖于转化含羟基分子。甘油就是这样的一个例子,它可以经过选择性的加氢脱氧反应产生丙二醇,这是一种有价值的化学前体。因此,甘油的加氢脱氧反应结合了直接的工业应用和与可持续原料相关的反应类基础研究的基础。考虑到大型有机分子的复杂性,大多数多相催化的建模工作都集中在小(C1-2)有机物的反应性上。甘油,以其c3 -主链为特征,有75种不同的气相构象,能量变化高达0.1 eV (Callam, C. S. J. Am)。化学。社会科学学报,2001,23(3):11743-11754。当考虑到它的11个反应键(C-O, C-H和O-H)时,甘油的反应性模型跨越了广泛的构象和反应空间。密度泛函理论模拟的高计算成本限制了对析因反应空间的详尽探索,导致对氢脱氧(HDO)机理的了解有限,并阻碍了合理的催化剂设计。因此,到目前为止,还没有系统的研究集中于全面采样甘油表面构象的能量学及其反应性。在本研究中,我们采用消息传递图神经网络架构(MACE)来开发机器学习力场(MLFF)电位,结合主动学习来研究构象复杂性对Cu(111)表面上甘油HDO反应网络的影响。经过5次迭代,我们训练的MLFF模型准确地预测了表面结合结构,均方根精度为0.04 eV (0.6 meV/原子总能量),这对于准确确定7条竞争路径上24个亚稳态和26个中间态的构象最小值至关重要。构象采样揭示了复杂能量景观的复杂本质,其中具有多个浅最小值的构象导致连接它们的过渡态能量的非平凡趋势。值得注意的是,与β-主链相比,研究预测具有α-和γ-主链的甘油结构的O-H键断裂的激活障碍较低。这在二级O-H甘油键断裂的情况下是显著的,其中根据初始甘油结构基序的不同,激活势垒变化高达0.44 eV。总之,我们确定脱氢-脱水-氢化是导致Cu(111)表面PDO形成的主要途径。甘油醛对C-H键断裂的选择性比C-OH键断裂的选择性解释了1,2- pdo比1,3- pdo更高的选择性。
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


