Tariku T Gudura, Hanny Sawaf, Randy Ogbenna, Ali Mehdi, Leal Herlitz, Surafel K Gebreselassie, Shane A Bobart
{"title":"The Clinical and Pathological Characteristics of Patients with Proliferative Glomerulonephritis with Monoclonal Immunoglobulin Deposits.","authors":"Tariku T Gudura, Hanny Sawaf, Randy Ogbenna, Ali Mehdi, Leal Herlitz, Surafel K Gebreselassie, Shane A Bobart","doi":"10.1159/000544864","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Proliferative glomerulonephritis with monoclonal immunoglobin (Ig) deposits (PGNMID) is a rare form of kidney disease associated with low monoclonal protein detection rates and poor renal outcomes. The lack of effective therapy is partly due to limited data and understanding of its pathogenesis.</p><p><strong>Methods: </strong>We conducted a retrospective analysis of 18 patients with PGNMID from the Cleveland Clinic Kidney Biopsy Epidemiology Project from January 2015 to March 2023.</p><p><strong>Results: </strong>PGNMID was more predominant among males (67%), and whites (78%), with median age of 60 years. Over 2/3rd of patients presented with hypertension, renal insufficiency, and hematuria, while 26% of patients had nephrotic syndrome. Mean serum creatinine and proteinuria at biopsy were 3.2 mg/dL and 4.3 g/g, respectively. A detectable monoclonal (M) protein was detected in 28% of cases; however, only 3 patients had underlying hematologic disorders (multiple myeloma, CLL, and B-cell lymphoma). An endocapillary hypercellularity/membranoproliferative pattern (72%), IgG/kappa (83%), and IgG3 (56%) were predominant findings on kidney pathology. Eight patients (44%) progressed to end-stage kidney disease (ESKD), with a median onset of 7.5 months after the initial kidney biopsy. While 6 patients achieved complete remission primarily with clone-directed therapy.</p><p><strong>Conclusion: </strong>Despite monoclonal protein deposition on kidney biopsy, 28% patients with PGNMID had a detectable monoclonal protein. Unlike other forms of paraproteinemic kidney diseases, renal outcomes for patients with PGNMID are poor, with 44% progressing to ESKD (median time 7.5 months) after kidney biopsy in our cohort. Clone-directed therapy to improve outcomes remains the mainstay of treatment, despite the absence of detectable clone in most cases. Thus, further investigation into the pathogenesis of PGNMID is warranted to guide future treatment discovery and clinical trials.</p>","PeriodicalId":73177,"journal":{"name":"Glomerular diseases","volume":"5 1","pages":"142-150"},"PeriodicalIF":0.0000,"publicationDate":"2025-02-20","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11936450/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Glomerular diseases","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000544864","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Proliferative glomerulonephritis with monoclonal immunoglobin (Ig) deposits (PGNMID) is a rare form of kidney disease associated with low monoclonal protein detection rates and poor renal outcomes. The lack of effective therapy is partly due to limited data and understanding of its pathogenesis.
Methods: We conducted a retrospective analysis of 18 patients with PGNMID from the Cleveland Clinic Kidney Biopsy Epidemiology Project from January 2015 to March 2023.
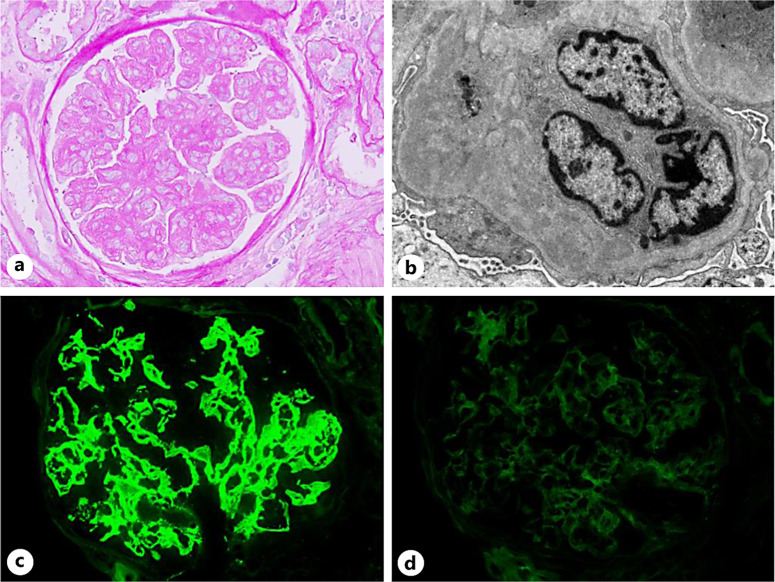
Results: PGNMID was more predominant among males (67%), and whites (78%), with median age of 60 years. Over 2/3rd of patients presented with hypertension, renal insufficiency, and hematuria, while 26% of patients had nephrotic syndrome. Mean serum creatinine and proteinuria at biopsy were 3.2 mg/dL and 4.3 g/g, respectively. A detectable monoclonal (M) protein was detected in 28% of cases; however, only 3 patients had underlying hematologic disorders (multiple myeloma, CLL, and B-cell lymphoma). An endocapillary hypercellularity/membranoproliferative pattern (72%), IgG/kappa (83%), and IgG3 (56%) were predominant findings on kidney pathology. Eight patients (44%) progressed to end-stage kidney disease (ESKD), with a median onset of 7.5 months after the initial kidney biopsy. While 6 patients achieved complete remission primarily with clone-directed therapy.
Conclusion: Despite monoclonal protein deposition on kidney biopsy, 28% patients with PGNMID had a detectable monoclonal protein. Unlike other forms of paraproteinemic kidney diseases, renal outcomes for patients with PGNMID are poor, with 44% progressing to ESKD (median time 7.5 months) after kidney biopsy in our cohort. Clone-directed therapy to improve outcomes remains the mainstay of treatment, despite the absence of detectable clone in most cases. Thus, further investigation into the pathogenesis of PGNMID is warranted to guide future treatment discovery and clinical trials.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


