Hayley A Ron, Owen Kane, Rose Guo, Caitlin Menello, Nicole Engelhardt, Shaney Pressley, Brenda DiBoscio, Madeline Steffensen, Sanmati Cuddapah, Kim Ng, Can Ficicioglu, Rebecca C Ahrens-Nicklas
{"title":"Five-Year Outcomes of Patients with Pompe Disease Identified by the Pennsylvania Newborn Screen.","authors":"Hayley A Ron, Owen Kane, Rose Guo, Caitlin Menello, Nicole Engelhardt, Shaney Pressley, Brenda DiBoscio, Madeline Steffensen, Sanmati Cuddapah, Kim Ng, Can Ficicioglu, Rebecca C Ahrens-Nicklas","doi":"10.3390/ijns11010016","DOIUrl":null,"url":null,"abstract":"<p><p>Pennsylvania started newborn screening for Pompe disease (PD) in 2016. As a result, the prevalence of PD has increased with early detection, primarily of late-onset Pompe disease (LOPD). No clear guidelines exist regarding if and when to initiate enzyme replacement therapy (ERT) in patients identified through a newborn screen (NBS). To help define the natural history and indications for starting ERT, we present the long-term follow-up data of 45 patients identified through NBS from 2016 to 2021. These patients were evaluated at regular intervals through our multi-disciplinary clinic at the Children's Hospital of Philadelphia (CHOP) with physical examinations, physical therapy evaluations, muscle biomarkers including creatine kinase (CK), aspartate aminotransferase (AST), alanine aminotransferase (ALT), and hexosaminidase 4 levels (Hex4), as well as cardiac evaluation at certain points in time. We found that newborn screening of acid alpha-glucosidase (GAA) enzyme detected primarily LOPD. One case of infantile-onset PD (IOPD) was detected. Muscle biomarkers in LOPD were elevated at birth and showed a general downward trend over time. NBS GAA levels and initial CK levels helped to differentiate LOPD cases from unaffected infants (carriers, pseudodeficiency alleles), while Hex4 was not a meaningful discriminator. On repeat NBS, there was a significant difference between mean GAA levels for the unaffected vs. compound heterozygote groups and unaffected vs. homozygote groups for the common splice site pathogenic variant (c.-32-13T>G). Echocardiogram and electrocardiogram (EKG) are essentially normal at the first evaluation in LOPD. One LOPD patient was started on ERT at age 4.5 months. Continued data collection on these patients is critical for developing management guidelines, including timing of ERT and improved genotype-phenotype correlation.</p>","PeriodicalId":14159,"journal":{"name":"International Journal of Neonatal Screening","volume":"11 1","pages":""},"PeriodicalIF":4.0000,"publicationDate":"2025-02-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11943203/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Neonatal Screening","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/ijns11010016","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
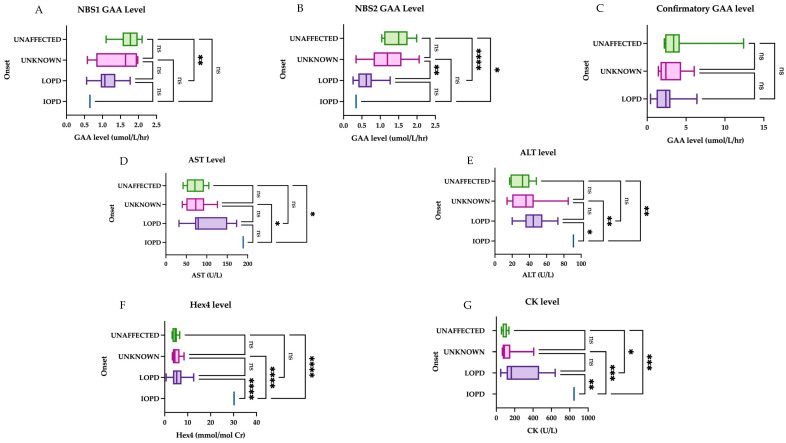
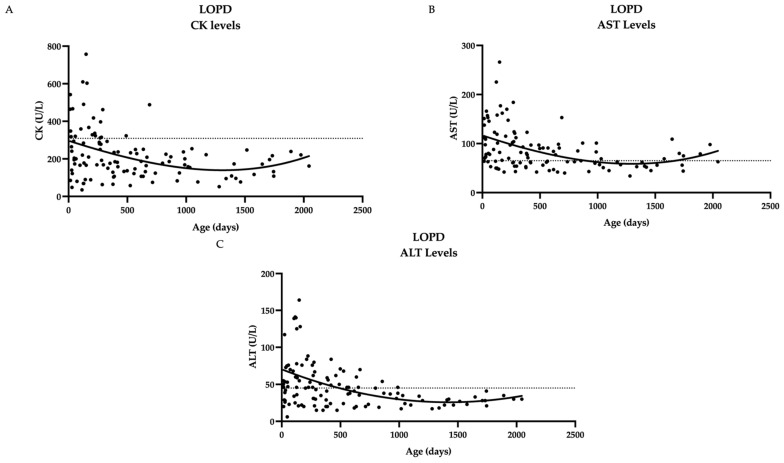
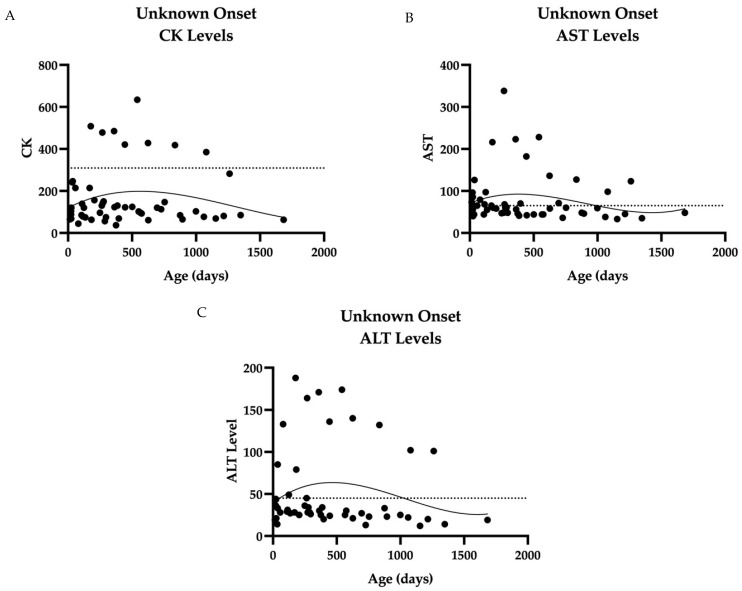
Pennsylvania started newborn screening for Pompe disease (PD) in 2016. As a result, the prevalence of PD has increased with early detection, primarily of late-onset Pompe disease (LOPD). No clear guidelines exist regarding if and when to initiate enzyme replacement therapy (ERT) in patients identified through a newborn screen (NBS). To help define the natural history and indications for starting ERT, we present the long-term follow-up data of 45 patients identified through NBS from 2016 to 2021. These patients were evaluated at regular intervals through our multi-disciplinary clinic at the Children's Hospital of Philadelphia (CHOP) with physical examinations, physical therapy evaluations, muscle biomarkers including creatine kinase (CK), aspartate aminotransferase (AST), alanine aminotransferase (ALT), and hexosaminidase 4 levels (Hex4), as well as cardiac evaluation at certain points in time. We found that newborn screening of acid alpha-glucosidase (GAA) enzyme detected primarily LOPD. One case of infantile-onset PD (IOPD) was detected. Muscle biomarkers in LOPD were elevated at birth and showed a general downward trend over time. NBS GAA levels and initial CK levels helped to differentiate LOPD cases from unaffected infants (carriers, pseudodeficiency alleles), while Hex4 was not a meaningful discriminator. On repeat NBS, there was a significant difference between mean GAA levels for the unaffected vs. compound heterozygote groups and unaffected vs. homozygote groups for the common splice site pathogenic variant (c.-32-13T>G). Echocardiogram and electrocardiogram (EKG) are essentially normal at the first evaluation in LOPD. One LOPD patient was started on ERT at age 4.5 months. Continued data collection on these patients is critical for developing management guidelines, including timing of ERT and improved genotype-phenotype correlation.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


