{"title":"Analysis of Common Alpha-Globin Gene Abnormalities and Their Effects as Genetic Modifiers in Thai Children With β-Globin Gene Abnormalities.","authors":"Sethapong Lertsakulbunlue, Boonchai Boonyawat, Chanchai Traivaree, Apichat Photia","doi":"10.1155/anem/9933808","DOIUrl":null,"url":null,"abstract":"<p><p>Beta-thalassemia exhibits a broad phenotypic range influenced by the severity of <i>HBB</i> mutation and various genetic modifiers. One of the most essential modifiers is the coinheritance of α-globin gene mutation. Nevertheless, the understanding of these α-globin variations' impact on beta-thalassemia is lacking among pediatric patients. This study investigated the impact of common α-globin gene mutations on clinical phenotype and hematological parameters in 122 Thai children with either β-thalassemia diseases or carriers recruited from Phramongkutklao Hospital, a major thalassemia center. Clinical characteristics, transfusion history, and hematological parameters were recorded, with molecular testing for common α-globin deletions and Hb CS mutations. The cohort included 8 homozygous β-thalassemia, 55 β-thalassemia/Hb E, 18 homozygous Hb E, 26 heterozygous Hb E, and 15 heterozygous β-thalassemia children. Coinheritance of α-globin mutations was less frequent in β-thalassemia diseases (6 of 63) than in β-thalassemia traits (25 of 59) (<i>p</i> < 0.001), indicating a potential modifier effect that reduces severity. Among β-thalassemia/Hb E patients, single α-globin deletions or Hb CS mutations were linked with lower Hb E, MCV, and MCH. Similarly, in both β-thalassemia and Hb E traits with α-globin gene mutation had significantly lower MCV, MCH and Hb E levels (only in the Hb E trait) and elevated RDW. Moreover, lower hematocrit and hemoglobin in these carriers were noted in cases coinherited with deletional Hb H disease initially undiagnosed by Hb typing. In conclusion, the diagnostic value of hematological parameters and Hb typing in identifying common α-globin mutations in pediatric β-thalassemia patients were highlighted. Hematological parameters are vital indicators that may prompt genetic screening to confirm α-globin abnormalities, supporting improved diagnosis and management of complex αβ-thalassemia syndromes.</p>","PeriodicalId":46055,"journal":{"name":"Anemia","volume":"2025 ","pages":"9933808"},"PeriodicalIF":2.6000,"publicationDate":"2025-03-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11932748/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Anemia","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1155/anem/9933808","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
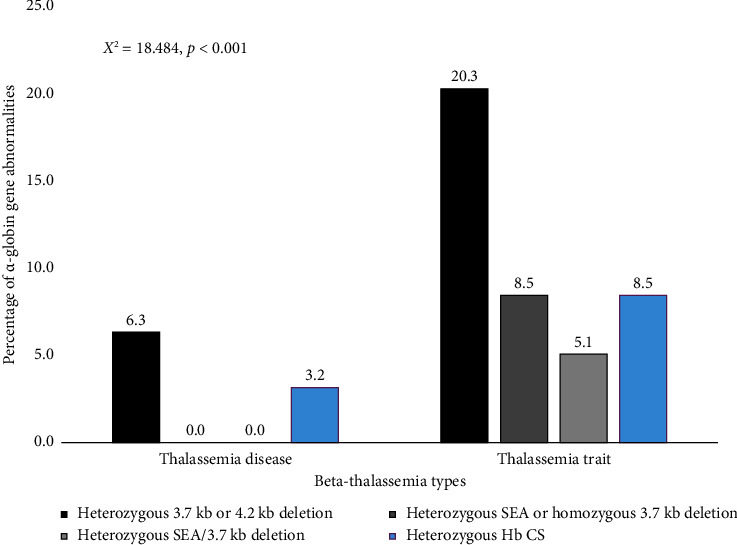
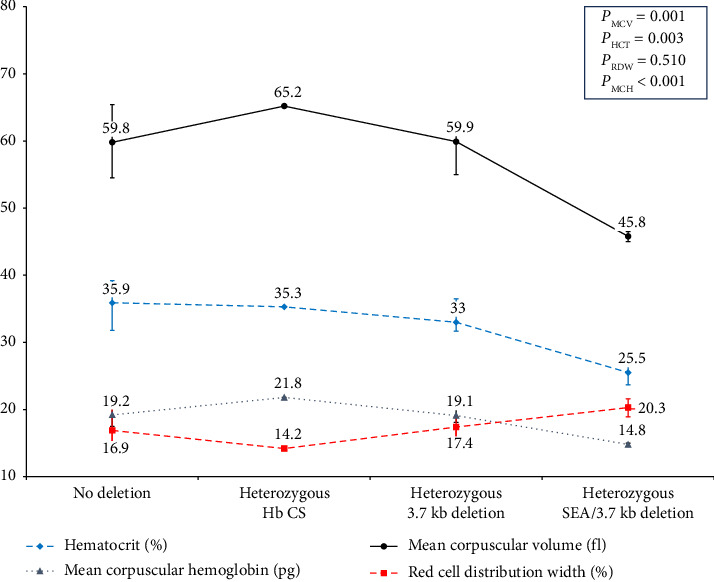
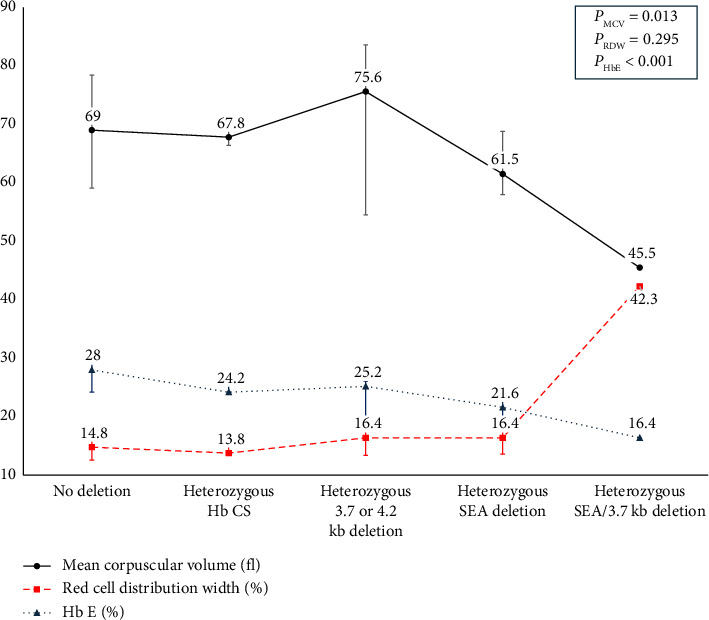
Beta-thalassemia exhibits a broad phenotypic range influenced by the severity of HBB mutation and various genetic modifiers. One of the most essential modifiers is the coinheritance of α-globin gene mutation. Nevertheless, the understanding of these α-globin variations' impact on beta-thalassemia is lacking among pediatric patients. This study investigated the impact of common α-globin gene mutations on clinical phenotype and hematological parameters in 122 Thai children with either β-thalassemia diseases or carriers recruited from Phramongkutklao Hospital, a major thalassemia center. Clinical characteristics, transfusion history, and hematological parameters were recorded, with molecular testing for common α-globin deletions and Hb CS mutations. The cohort included 8 homozygous β-thalassemia, 55 β-thalassemia/Hb E, 18 homozygous Hb E, 26 heterozygous Hb E, and 15 heterozygous β-thalassemia children. Coinheritance of α-globin mutations was less frequent in β-thalassemia diseases (6 of 63) than in β-thalassemia traits (25 of 59) (p < 0.001), indicating a potential modifier effect that reduces severity. Among β-thalassemia/Hb E patients, single α-globin deletions or Hb CS mutations were linked with lower Hb E, MCV, and MCH. Similarly, in both β-thalassemia and Hb E traits with α-globin gene mutation had significantly lower MCV, MCH and Hb E levels (only in the Hb E trait) and elevated RDW. Moreover, lower hematocrit and hemoglobin in these carriers were noted in cases coinherited with deletional Hb H disease initially undiagnosed by Hb typing. In conclusion, the diagnostic value of hematological parameters and Hb typing in identifying common α-globin mutations in pediatric β-thalassemia patients were highlighted. Hematological parameters are vital indicators that may prompt genetic screening to confirm α-globin abnormalities, supporting improved diagnosis and management of complex αβ-thalassemia syndromes.
期刊介绍:
Anemia is a peer-reviewed, Open Access journal that publishes original research articles, review articles, and clinical studies on all types of anemia. Articles focusing on patient care, health systems, epidemiology, and animal models will be considered, among other relevant topics. Affecting roughly one third of the world’s population, anemia is a major public health concern. The journal aims to facilitate the exchange of research addressing global health and mortality relating to anemia and associated diseases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


