Zsófia Flóra Nagy, György Pfliegler, János Kósa, Kristóf Árvai, Ildikó Istenes, Attila Doros, Botond Timár, Péter Lakatos, Judit Demeter
{"title":"Case Report: Importance of high-throughput genetic investigations in the differential diagnosis of unexplained erythrocytosis.","authors":"Zsófia Flóra Nagy, György Pfliegler, János Kósa, Kristóf Árvai, Ildikó Istenes, Attila Doros, Botond Timár, Péter Lakatos, Judit Demeter","doi":"10.3389/pore.2025.1612037","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Polycythemia indicates the pathological increase in the number of red blood cells and the rise of hematocrit values. Polyglobulia can be of primary or secondary origin, with the most common primary polycythemia being a myeloproliferative neoplasm, polycythemia vera. Polyglobulia patients may develop cardiovascular complications and thromboembolic events. The gold standard of first-line treatment in polycythemia vera is phlebotomy, which is indicated to keep the hematocrit value below 0.45. Until now the goal to be achieved in secondary polyglobulia has been similar. In secondary polyglobulia this rule of thumb needs to be re-evaluated as shown by the example of two patients suffering from different rare, genetically determined polyglobulias. In our article we present the case of these two patients and discuss the diagnostic and therapeutic principles to be applied in patients with rare, genetically determined polyglobulias.</p><p><strong>Patients and methods: </strong>After completing the usual diagnostic algorithm for polyglobulia no cause could be identified in two of our male patients. Therefore, we set out to perform whole exome sequencing in both patients. Our analysis did not include copy number analysis.</p><p><strong>Results: </strong>In Patient 1 the p.Ser179Pro variant in the <i>VHL</i> gene was detected in the homozygous state, which is classified as likely pathogenic according to the ACMG guidelines. Homozygous <i>VHL</i> mutations are implicated in Chuvash polycythemia. Segregation analysis was declined by the family. In Patient 2 the <i>PKLR</i> gene p.His306Gln variant was detected in the heterozygous form. The gene plays a role in pyruvate metabolism. Family screening did not detect this variant in healthy family members.</p><p><strong>Discussion: </strong>We identified rare, possibly pathogenic genetic variants in two patients with polyglobulia and as a consequence of the genetic diagnosis we implemented individualized patient monitoring. We recommend the utilization of high-throughput genomic testing in cases with unexplained polyglobulia.</p>","PeriodicalId":19981,"journal":{"name":"Pathology & Oncology Research","volume":"31 ","pages":"1612037"},"PeriodicalIF":2.3000,"publicationDate":"2025-03-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11930660/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pathology & Oncology Research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.3389/pore.2025.1612037","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q3","JCRName":"ONCOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Polycythemia indicates the pathological increase in the number of red blood cells and the rise of hematocrit values. Polyglobulia can be of primary or secondary origin, with the most common primary polycythemia being a myeloproliferative neoplasm, polycythemia vera. Polyglobulia patients may develop cardiovascular complications and thromboembolic events. The gold standard of first-line treatment in polycythemia vera is phlebotomy, which is indicated to keep the hematocrit value below 0.45. Until now the goal to be achieved in secondary polyglobulia has been similar. In secondary polyglobulia this rule of thumb needs to be re-evaluated as shown by the example of two patients suffering from different rare, genetically determined polyglobulias. In our article we present the case of these two patients and discuss the diagnostic and therapeutic principles to be applied in patients with rare, genetically determined polyglobulias.
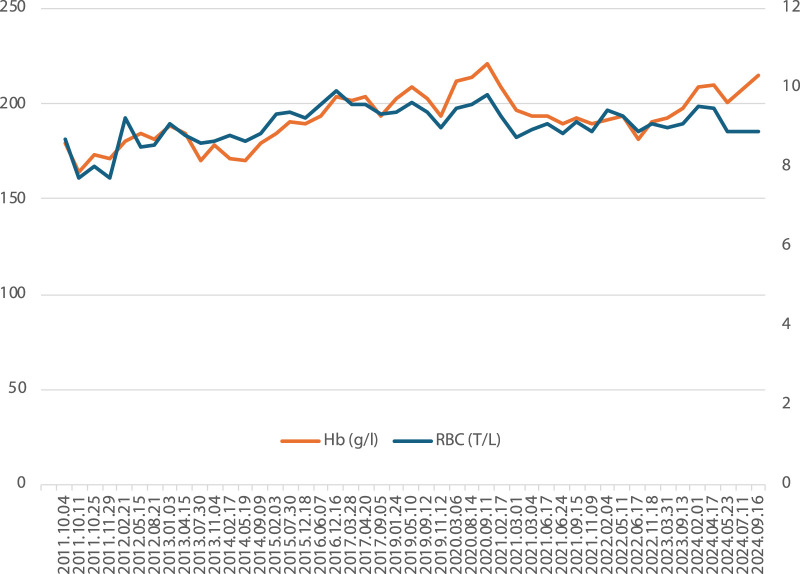
Patients and methods: After completing the usual diagnostic algorithm for polyglobulia no cause could be identified in two of our male patients. Therefore, we set out to perform whole exome sequencing in both patients. Our analysis did not include copy number analysis.
Results: In Patient 1 the p.Ser179Pro variant in the VHL gene was detected in the homozygous state, which is classified as likely pathogenic according to the ACMG guidelines. Homozygous VHL mutations are implicated in Chuvash polycythemia. Segregation analysis was declined by the family. In Patient 2 the PKLR gene p.His306Gln variant was detected in the heterozygous form. The gene plays a role in pyruvate metabolism. Family screening did not detect this variant in healthy family members.
Discussion: We identified rare, possibly pathogenic genetic variants in two patients with polyglobulia and as a consequence of the genetic diagnosis we implemented individualized patient monitoring. We recommend the utilization of high-throughput genomic testing in cases with unexplained polyglobulia.
期刊介绍:
Pathology & Oncology Research (POR) is an interdisciplinary Journal at the interface of pathology and oncology including the preclinical and translational research, diagnostics and therapy. Furthermore, POR is an international forum for the rapid communication of reviews, original research, critical and topical reports with excellence and novelty. Published quarterly, POR is dedicated to keeping scientists informed of developments on the selected biomedical fields bridging the gap between basic research and clinical medicine. It is a special aim for POR to promote pathological and oncological publishing activity of colleagues in the Central and East European region. The journal will be of interest to pathologists, and a broad range of experimental and clinical oncologists, and related experts. POR is supported by an acknowledged international advisory board and the Arányi Fundation for modern pathology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


