Konstantinos Kordos, Konstantinos Kaklamanis, Maria Andrea, Dimitrios G Papageorgiou
{"title":"PCDTBT: Force Field Parameterization and Properties by Molecular Dynamics Simulation.","authors":"Konstantinos Kordos, Konstantinos Kaklamanis, Maria Andrea, Dimitrios G Papageorgiou","doi":"10.1021/acs.jpcb.4c08393","DOIUrl":null,"url":null,"abstract":"<p><p>Conjugated polymers are indispensable building blocks in a variety of organic electronics applications such as solar cells, light-emitting diodes, and field-effect transistors. Poly[<i>N</i>-9'-heptadecanyl-2,7-carbazole-<i>alt</i>-5,5-(4',7'-di-2-thienyl-2',1',3'-benzothiadiazole)] (PCDTBT) is a carbazole-benzothiadiazole-based copolymer with a donor-acceptor structure, consisting of electron-donating and electron-withdrawing subunits and featuring a low band gap. In this work, the General Amber Force Field is extended in two ways, specifically for modeling PCDTBT. First, a set of partial atomic charges is derived that mimic a long chain and adequately describe different conformations that may be encountered in a bulk environment. Second, torsional terms are reparametrized for all dihedral angles in the backbone via ab initio computations. Subsequently, a series of large-scale Molecular Dynamics simulations are employed to construct and equilibrate bulk ensembles of three PCDTBT oligomers using different starting conformations of the oligomer chains. Several structural properties are computed, namely mass density, chain stiffness (through persistence length and Kuhn segment length), and glass transition temperature. Our results are in good agreement with available literature data, demonstrating the suitability of the new parametrization.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":" ","pages":"3492-3501"},"PeriodicalIF":2.9000,"publicationDate":"2025-04-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11973877/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcb.4c08393","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/3/20 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
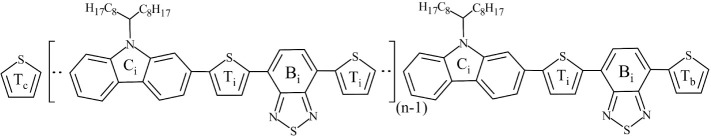
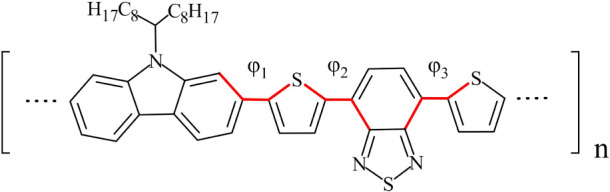
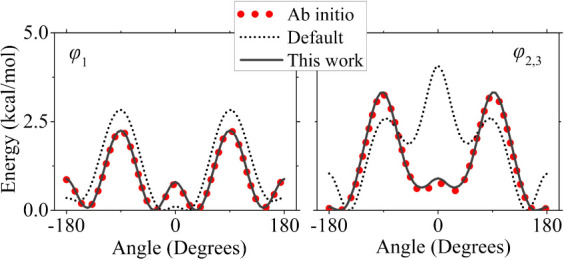
Conjugated polymers are indispensable building blocks in a variety of organic electronics applications such as solar cells, light-emitting diodes, and field-effect transistors. Poly[N-9'-heptadecanyl-2,7-carbazole-alt-5,5-(4',7'-di-2-thienyl-2',1',3'-benzothiadiazole)] (PCDTBT) is a carbazole-benzothiadiazole-based copolymer with a donor-acceptor structure, consisting of electron-donating and electron-withdrawing subunits and featuring a low band gap. In this work, the General Amber Force Field is extended in two ways, specifically for modeling PCDTBT. First, a set of partial atomic charges is derived that mimic a long chain and adequately describe different conformations that may be encountered in a bulk environment. Second, torsional terms are reparametrized for all dihedral angles in the backbone via ab initio computations. Subsequently, a series of large-scale Molecular Dynamics simulations are employed to construct and equilibrate bulk ensembles of three PCDTBT oligomers using different starting conformations of the oligomer chains. Several structural properties are computed, namely mass density, chain stiffness (through persistence length and Kuhn segment length), and glass transition temperature. Our results are in good agreement with available literature data, demonstrating the suitability of the new parametrization.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


