CO2 Reduction Mechanism Based on a Partially Reduced Cu Single-Atom Catalyst: A First-Principles Study
IF 3.2
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
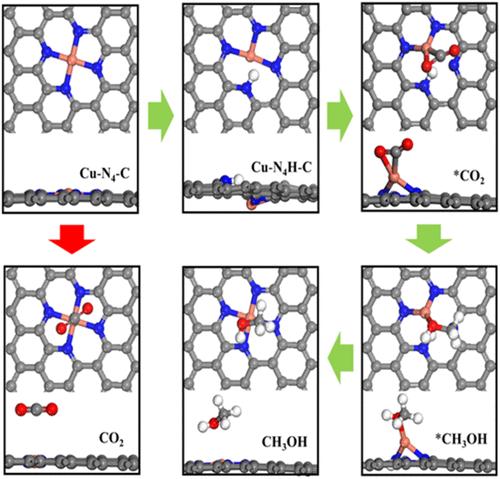
Experimentally, CO can be generated as the main product at a lower negative potential. Since CO is the key intermediate of methanol synthesis and C–C coupling reactions, the catalyst should have a strong adsorption capacity for CO molecules. Theoretical computations, based on Cu–N4–C models, indicate that a significantly high-energy barrier needs to be for carboxylation of CO2, resulting primarily in formic acid formation and exhibits weak adsorption of CO. Given the abundance of N–H bonds involved in both the preparation of N@graphene and electrocatalytic reduction, a Cu–N4H–C structural model has been proposed, and the model can remain stable at 600 K up to 3 ps based on AIMD simulations. In the Cu–N4H–C model, the Cu+ ion is partially reduced and enhances the adsorption capacity of CO. The model effectively lowers the energy barrier for CO2 carboxylation to 0.23 eV, and the energy barrier for methanol synthesis is 0.50 eV. By doping with K atoms, the catalyst can also be reduced. The reduced catalyst shows an ideal reduction performance of CO2 to HCOOH. Based on ab initio simulations, this study will reveal the influence of the microenvironment on the CO2 reduction process in Cu single-atom catalysts and is helpful for the realization of efficient CO2 reduction in experiments.
基于部分还原Cu单原子催化剂的CO2还原机理:第一性原理研究
实验表明,CO可以在较低的负电位下作为主要产物生成。由于CO是甲醇合成和C-C偶联反应的关键中间体,催化剂对CO分子应具有较强的吸附能力。基于Cu-N4-C模型的理论计算表明,CO的羧基化需要一个显著的高能屏障,主要导致甲酸的形成,并表现出对CO的弱吸附。考虑到制备N@graphene和电催化还原过程中都涉及到N-H键的丰富度,我们提出了一个Cu-N4-C结构模型,该模型可以在600 K至3 ps的条件下保持稳定。在Cu - n4h - c模型中,Cu+离子被部分还原,提高了CO的吸附能力,该模型有效地将CO2羧化的能垒降低到0.23 eV,甲醇合成的能垒为0.50 eV。通过掺杂K原子,催化剂也可以被还原。还原后的催化剂具有理想的将CO2还原为HCOOH的性能。本研究通过从头算模拟,揭示微环境对Cu单原子催化剂中CO2还原过程的影响,有助于实验中实现高效的CO2还原。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


