{"title":"Deficiency of Adenosine Deaminase 2 Masquerading as Behçet's Disease: Phenotypic Mimicry with HLA-B*51 Positivity.","authors":"Abdullah Almojali, Abdulrahman Alrasheed, Bushra Alharbi, Reem Alharbi, Wafaa Alsuwairi, Fayhan Alroqi, Jubran Alqanatish","doi":"10.1007/s10875-025-01876-0","DOIUrl":null,"url":null,"abstract":"<p><strong>Purpose: </strong>Deficiency of adenosine deaminase 2 (DADA2) is a rare monogenic autoinflammatory disease resulting from biallelic loss-of-function mutations in ADA2 gene. It has variable clinical manifestations, some of which can mimic Behçet's disease (BD). Herein, we present a family of three siblings diagnosed with DADA2, two of whom were initially misdiagnosed as BD based on clinical phenotype including positive human leukocyte antigen B51 (HLA-B*51).</p><p><strong>Methods: </strong>Gene mutational analysis was performed by whole exome (WES) and Sanger sequencing.</p><p><strong>Results: </strong>We reported two siblings presented with recurrent oral ulcers, fever, arthritis, and skin lesions, alongside elevated inflammatory markers and HLA-B*51 positivity, leading to an initial misdiagnosis of BD. Genetic testing later revealed a homozygous ADA2 variant (c.139G > A p.Gly47Arg) in both siblings and their asymptomatic younger sister, confirming DADA2 diagnosis. Thereafter, we reviewed the literature to identify other patients misdiagnosed with BD but later found to have DADA2. This resulted in a cohort of 10 DADA2 patients, including our two reported siblings. The median time from symptoms onset to the final diagnosis of DADA2 was 7 years. All patients exhibited BD-like phenotype, except for uveitis, and 8 were HLA-B*51 positive, which likely contributed to the diagnostic confusion.</p><p><strong>Conclusion: </strong>These findings highlight the broad clinical spectrum of DADA2, which can resemble BD, and suggest that HLA-B*51 positivity in DADA2 may further complicate diagnosis. Clinicians should maintain a high index of suspicion for DADA2 in early-onset BD-like cases, particularly without uveitis, or a family history of similar symptoms. Further studies are warranted to explore HLA-B*51 role in DADA2 phenotype.</p>","PeriodicalId":15531,"journal":{"name":"Journal of Clinical Immunology","volume":"45 1","pages":"83"},"PeriodicalIF":5.7000,"publicationDate":"2025-03-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11913947/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Clinical Immunology","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1007/s10875-025-01876-0","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"IMMUNOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Purpose: Deficiency of adenosine deaminase 2 (DADA2) is a rare monogenic autoinflammatory disease resulting from biallelic loss-of-function mutations in ADA2 gene. It has variable clinical manifestations, some of which can mimic Behçet's disease (BD). Herein, we present a family of three siblings diagnosed with DADA2, two of whom were initially misdiagnosed as BD based on clinical phenotype including positive human leukocyte antigen B51 (HLA-B*51).
Methods: Gene mutational analysis was performed by whole exome (WES) and Sanger sequencing.
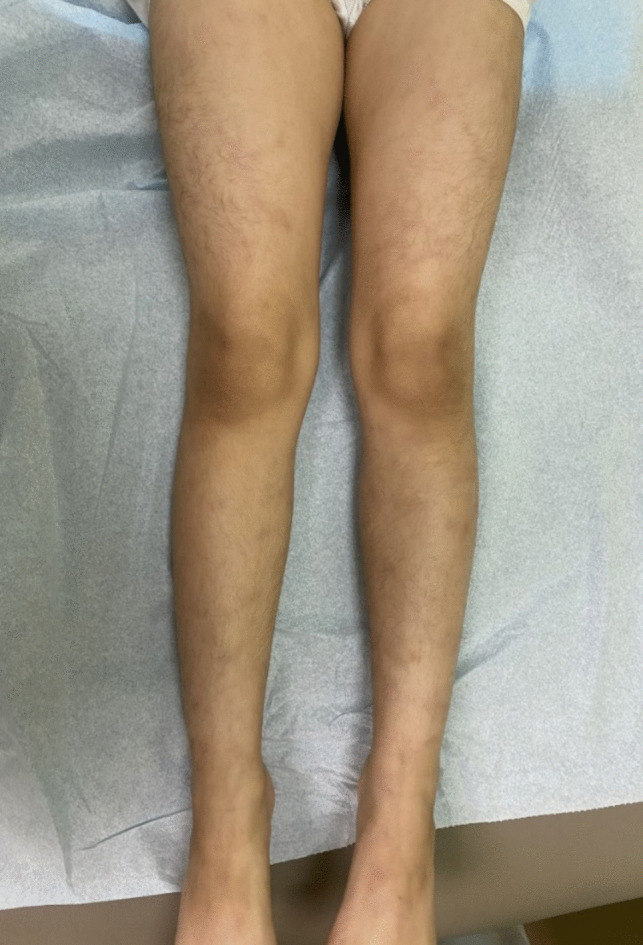
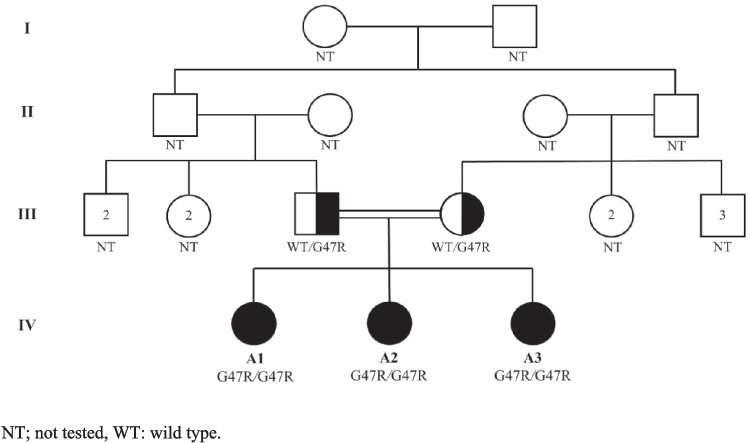
Results: We reported two siblings presented with recurrent oral ulcers, fever, arthritis, and skin lesions, alongside elevated inflammatory markers and HLA-B*51 positivity, leading to an initial misdiagnosis of BD. Genetic testing later revealed a homozygous ADA2 variant (c.139G > A p.Gly47Arg) in both siblings and their asymptomatic younger sister, confirming DADA2 diagnosis. Thereafter, we reviewed the literature to identify other patients misdiagnosed with BD but later found to have DADA2. This resulted in a cohort of 10 DADA2 patients, including our two reported siblings. The median time from symptoms onset to the final diagnosis of DADA2 was 7 years. All patients exhibited BD-like phenotype, except for uveitis, and 8 were HLA-B*51 positive, which likely contributed to the diagnostic confusion.
Conclusion: These findings highlight the broad clinical spectrum of DADA2, which can resemble BD, and suggest that HLA-B*51 positivity in DADA2 may further complicate diagnosis. Clinicians should maintain a high index of suspicion for DADA2 in early-onset BD-like cases, particularly without uveitis, or a family history of similar symptoms. Further studies are warranted to explore HLA-B*51 role in DADA2 phenotype.
目的:腺苷脱氨酶2缺乏症(DADA2)是一种罕见的单基因自身炎症性疾病,由ADA2基因双等位基因功能丧失突变引起。它具有多种临床表现,其中一些可以模仿behaperet病(BD)。在此,我们报告了一个被诊断为DADA2的三兄弟姐妹家庭,其中两人最初被误诊为BD,基于临床表型包括阳性的人白细胞抗原B51 (HLA-B*51)。方法:采用全外显子组(WES)和Sanger测序进行基因突变分析。结果:我们报告了两名兄弟姐妹出现复发性口腔溃疡、发烧、关节炎和皮肤病变,同时炎症标志物升高和HLA-B*51阳性,导致最初误诊为双相障碍。随后的基因检测在两名兄弟姐妹及其无症状的妹妹中发现了ADA2纯合子变体(c.139G > a p.Gly47Arg),证实了DADA2的诊断。此后,我们回顾了文献,以确定其他被误诊为BD但后来被发现患有DADA2的患者。这导致了10名DADA2患者的队列,包括我们报告的两位兄弟姐妹。从症状出现到最终诊断DADA2的中位时间为7年。除葡萄膜炎外,所有患者均表现为bd样表型,其中8例HLA-B*51阳性,这可能导致诊断混乱。结论:这些发现突出了DADA2的广泛临床谱,它可能类似于BD,提示HLA-B*51阳性的DADA2可能进一步复杂化诊断。对于早发性bd样病例,临床医生应保持对DADA2的高度怀疑,特别是没有葡萄膜炎或有类似症状家族史的病例。HLA-B*51在DADA2表型中的作用有待进一步研究。
期刊介绍:
The Journal of Clinical Immunology publishes impactful papers in the realm of human immunology, delving into the diagnosis, pathogenesis, prognosis, or treatment of human diseases. The journal places particular emphasis on primary immunodeficiencies and related diseases, encompassing inborn errors of immunity in a broad sense, their underlying genotypes, and diverse phenotypes. These phenotypes include infection, malignancy, allergy, auto-inflammation, and autoimmunity. We welcome a broad spectrum of studies in this domain, spanning genetic discovery, clinical description, immunologic assessment, diagnostic approaches, prognosis evaluation, and treatment interventions. Case reports are considered if they are genuinely original and accompanied by a concise review of the relevant medical literature, illustrating how the novel case study advances the field. The instructions to authors provide detailed guidance on the four categories of papers accepted by the journal.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


