Andreas Luttens, Israel Cabeza de Vaca, Leonard Sparring, José Brea, Antón Leandro Martínez, Nour Aldin Kahlous, Dmytro S. Radchenko, Yurii S. Moroz, María Isabel Loza, Ulf Norinder, Jens Carlsson
{"title":"Rapid traversal of vast chemical space using machine learning-guided docking screens","authors":"Andreas Luttens, Israel Cabeza de Vaca, Leonard Sparring, José Brea, Antón Leandro Martínez, Nour Aldin Kahlous, Dmytro S. Radchenko, Yurii S. Moroz, María Isabel Loza, Ulf Norinder, Jens Carlsson","doi":"10.1038/s43588-025-00777-x","DOIUrl":null,"url":null,"abstract":"The accelerating growth of make-on-demand chemical libraries provides unprecedented opportunities to identify starting points for drug discovery with virtual screening. However, these multi-billion-scale libraries are challenging to screen, even for the fastest structure-based docking methods. Here we explore a strategy that combines machine learning and molecular docking to enable rapid virtual screening of databases containing billions of compounds. In our workflow, a classification algorithm is trained to identify top-scoring compounds based on molecular docking of 1 million compounds to the target protein. The conformal prediction framework is then used to make selections from the multi-billion-scale library, reducing the number of compounds to be scored by docking. The CatBoost classifier showed an optimal balance between speed and accuracy and was used to adapt the workflow for screens of ultralarge libraries. Application to a library of 3.5 billion compounds demonstrated that our protocol can reduce the computational cost of structure-based virtual screening by more than 1,000-fold. Experimental testing of predictions identified ligands of G protein-coupled receptors and demonstrated that our approach enables discovery of compounds with multi-target activity tailored for therapeutic effect. Combining conformal prediction machine learning with molecular docking, a method to efficiently screen multi-billion-scale libraries is developed, enabling the discovery of a dual-target ligand modulating the A2A adenosine and D2 dopamine receptors.","PeriodicalId":74246,"journal":{"name":"Nature computational science","volume":"5 4","pages":"301-312"},"PeriodicalIF":18.3000,"publicationDate":"2025-03-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12021657/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature computational science","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s43588-025-00777-x","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
引用次数: 0
Abstract
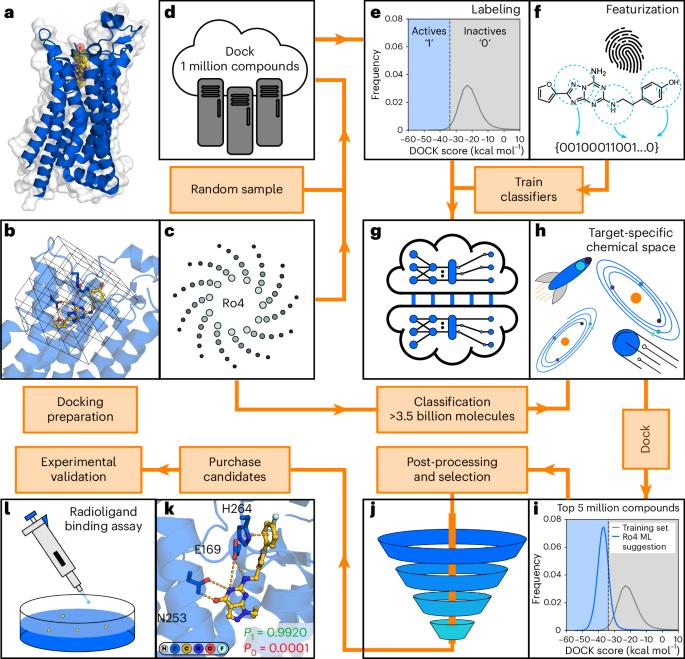
The accelerating growth of make-on-demand chemical libraries provides unprecedented opportunities to identify starting points for drug discovery with virtual screening. However, these multi-billion-scale libraries are challenging to screen, even for the fastest structure-based docking methods. Here we explore a strategy that combines machine learning and molecular docking to enable rapid virtual screening of databases containing billions of compounds. In our workflow, a classification algorithm is trained to identify top-scoring compounds based on molecular docking of 1 million compounds to the target protein. The conformal prediction framework is then used to make selections from the multi-billion-scale library, reducing the number of compounds to be scored by docking. The CatBoost classifier showed an optimal balance between speed and accuracy and was used to adapt the workflow for screens of ultralarge libraries. Application to a library of 3.5 billion compounds demonstrated that our protocol can reduce the computational cost of structure-based virtual screening by more than 1,000-fold. Experimental testing of predictions identified ligands of G protein-coupled receptors and demonstrated that our approach enables discovery of compounds with multi-target activity tailored for therapeutic effect. Combining conformal prediction machine learning with molecular docking, a method to efficiently screen multi-billion-scale libraries is developed, enabling the discovery of a dual-target ligand modulating the A2A adenosine and D2 dopamine receptors.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


